Introduction
Alglucosidase Alfa is a biosimilar drug that has been developed as a treatment for Pompe disease, a rare genetic disorder caused by a deficiency of the enzyme acid alpha-glucosidase (GAA). This enzyme is responsible for breaking down glycogen, a complex sugar molecule, in the body. Without enough GAA, glycogen builds up in the cells and tissues, leading to progressive muscle weakness and other symptoms. Alglucosidase Alfa is a recombinant form of human GAA, produced through genetic engineering techniques, and is used to replace the missing enzyme in patients with Pompe disease.
Structure of Alglucosidase Alfa
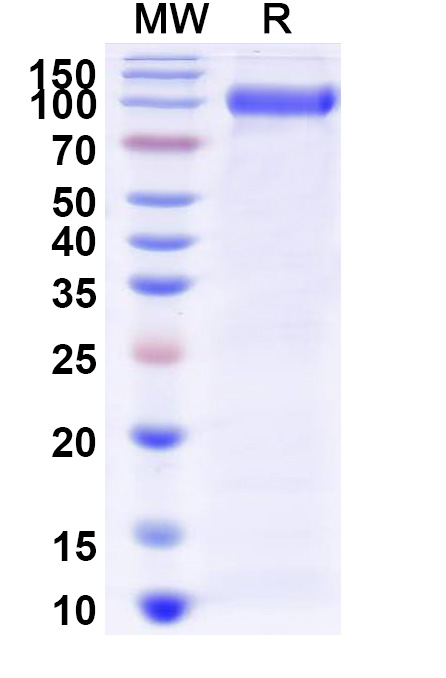
Alglucosidase Alfa is a glycoprotein, meaning it is composed of both protein and carbohydrate components. The protein component is a single chain of 952 amino acids, with a molecular weight of 99 kDa. The carbohydrate component is made up of complex sugar chains, which are important for the stability and function of the protein. Alglucosidase Alfa is produced in a mammalian cell expression system, ensuring that it is structurally and functionally similar to the human GAA enzyme.
Mechanism of Action
Alglucosidase Alfa works by replacing the missing or deficient GAA enzyme in patients with Pompe disease. It is administered intravenously and is taken up by cells throughout the body, where it breaks down glycogen into smaller, more manageable molecules. This helps to prevent the buildup of glycogen in various tissues and organs, particularly in the muscles. By reducing glycogen accumulation, Alglucosidase Alfa can slow down the progression of muscle weakness and other symptoms of Pompe disease.
Therapeutic Target
The therapeutic target of Alglucosidase Alfa is the enzyme acid alpha-glucosidase (GAA). This enzyme is found in the lysosomes, which are small compartments within cells that are responsible for breaking down and recycling various molecules. In patients with Pompe disease, mutations in the GAA gene result in a deficiency or complete absence of the GAA enzyme, leading to the buildup of glycogen in the lysosomes. By targeting this enzyme, Alglucosidase Alfa can help to restore the normal function of the lysosomes and prevent glycogen accumulation.
Application of Alglucosidase Alfa
Alglucosidase Alfa is indicated for the treatment of Pompe disease in both pediatric and adult patients. It is administered through intravenous infusion every two weeks and has been shown to improve muscle strength and function, as well as respiratory function, in patients with Pompe disease. Alglucosidase Alfa has been approved by regulatory bodies in various countries, including the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA). It is also available as a biosimilar in some countries, providing a more affordable option for patients.
Conclusion
Alglucosidase Alfa is a biosimilar drug that has been developed as a treatment for Pompe disease. It is a recombinant form of the human GAA enzyme, produced through genetic engineering techniques. By targeting the deficient GAA enzyme, Alglucosidase Alfa helps to reduce glycogen accumulation and improve muscle and respiratory function in patients with Pompe disease. It has been approved for use in both pediatric and adult patients and is available as a biosimilar in some countries, providing a more cost-effective treatment option.
Keywords
Alglucosidase Alfa, biosimilar, Pompe disease, acid alpha-glucosidase, enzyme, glycogen, muscle weakness, lysosomes, intravenous infusion, pediatric, adult, regulatory bodies, affordable.
There are no reviews yet.