
Cart (0 Items)
Your cart is currently empty.
View ProductsIt looks like you are visiting from outside the EU. Switch to the US version to see local pricing in USD and local shipping.
Switch to US ($) Antibody-drug conjugates
Antibody-drug conjugates
 Thomas Meyer
Thomas Meyer
Antibody-drug conjugates (ADCs) are an emerging class of biopharmaceuticals that have found great applicability in the treatment of oncological diseases. ADCs combine the specificity of monoclonal antibodies with the high toxicity of small drugs to produce a safer alternative to conventional chemotherapy.
As of July 2021, at least 10 ADCs have been approved by the FDA and EMA, 60% of these have reached the market only from 2019 onward. This recent boost in approvals can be explained by the significant improvements in linker chemistry achieved in the past decade. The active field of research is expected to make ADCs more stable and easier to produce which, in turn, might expand their applicability beyond anticancer therapies.
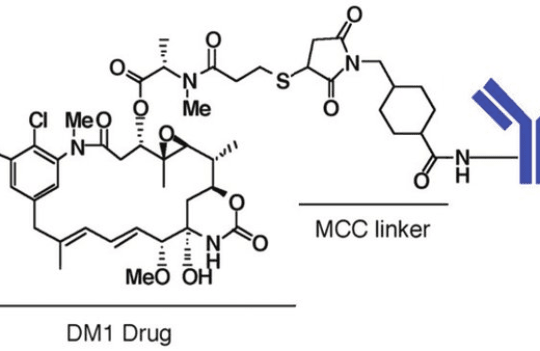
Linkers can be defined as the interface of chemistry (drug) and biology (antibody) in ADCs. Through a chemical bond, linkers bind small cytotoxic cargoes to large antibodies with the intent of increasing the selectivity of chemotherapeutics. Linkers consist of two main domains: (i) an antibody-binding domain and (ii) a payload-binding domain.
Linkers are a vital component of ADCs since they dictate the following physicochemical properties of these conjugates:
Before the recent boost in approvals, conventional ADCs had always been considered unstable and unreliable which considerably hindered their development. At that time, the number of chemical linkers available on the ADC toolbox was considerably smaller and many were known to degrade, albeit at slow rates, under physiological conditions and far from tumor sites. The high off-target toxicity continues to be one of the major challenges when it comes to ADC development.
The subsequential improvement of linker chemistry took place once researchers started experimenting with different molecules, adding spacers for steric protection, and leveraging substitution reactions to ensure linker stability. Today, many new linkers have been described and can be generally divided into two main classes: cleavable and non-cleavable linkers.
Chemically cleavable and enzyme cleavable linkers comprise the two major subclasses of cleavable linkers. This type of chemistry is defined by its susceptibility to hydrolysis under specific conditions or chemical signals. The incorporated chemical trigger restricts the cleavage and subsequent payload release to the intracellular environment or necrotic tissues.
The vast majority of ADCs currently on the market employ this type of chemistry and the two major motifs are peptides and disulfides. Peptide motifs, particularly the ones containing Val-Cit (valine and citrulline), have shown great stability and plasma and fast cleavage in the presence of intracellular proteases like cathepsin B. In contrast, disulfide motifs have been conventionally used with hydrazones to produce chemically labile bonds susceptible to hydrolysis in lysosomal compartments (low pH).
Alternative linkers like β-glucuronide, β-galactoside linkers, and PEGylated linkers have been gaining traction over the more conventional technologies due to their enhanced hydrophilicity which subsequently leads to a lower aggregation and higher plasma stability.
Non-cleavable linkers are defined by their unique mechanism of action. These molecules are stable and never separate from their defined payloads even upon ADC internalization. Contrary to their cleavable counterparts, ADCs with non-cleavable linkers need to be internalized by their target cells and the carrier antibody needs to be processed in the lysosome for linker-payload release to occur. This implies that the linker significantly changes the conformation and activity of the payload, but it also guarantees the optimal safety of these conjugates as the risk of premature payload release is minimized.
The research and development of new non-cleavable linker chemistries have lagged in comparison to cleavable chemistries. To date, only two ADCs using this type of bond have been approved – ado-trastuzumab emtansine and belantamab mafodotin. The first employs an SMCC linker and the second an MC linker, collectively known as thioether linkers.
Summary of the main types of cleavable and non-cleavable linkers used in ADC development.
| Linker chemistry | Subclass | Groups | Examples | FDA or EMA approved ADCs |
|---|---|---|---|---|
| Cleavable | Acid cleavable linkers | Hydrazone-disulfides | 4-(4′-acetylphenoxy) butanoic acid (acetyl butyrate) linker [AcBut linker] | Gemtuzumab ozogamicin; Inotuzumab ozogamicin |
| PEGylated | PEG8- and triazole-containing PABC-peptide-mc linker [CL2A linker] | Sacituzumab govitecan | ||
| Enzyme cleavable linkers | Peptide linkers | Valine-Citrulline [Val-Cit linker]
Valine-Alanine [Val-Ala linker] |
Brentuximab vedotin; Polatuzumab vedotin; Loncastuximab tesirine | |
| Noncleavable linkers | – | Thioether linkers | Succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate linker [SMCC linker]
Maleimidocaproyl linker [MC linker] |
Belantamab mafodotin; Ado-trastuzumab emtansine |
Each type of linker chemistry has its advantages and limitations. However, as a general rule, the choice of a linker should depend on the following properties:
In sum, choosing the proper linker chemistry starts by understanding the mechanisms of disease and testing different linker chemistries to obtain an optimal balance between safety and therapeutic efficiency.
One of the greatest advantages of using antibodies for cytotoxic drug delivery is their long half-life in circulation (between 11-30 days). However, this quickly becomes a hindrance when linkers show poor stability in the same conditions. Many of the early linker chemistries were quickly hydrolyzed in plasma (in a few hours) making them suitable for ADC applications.
This instability was shown to correlate with several factors:
Although the vast majority of linkers currently approved are protease-cleavable, the variety of linkers available for conjugation are expected to increase. Recently, several research groups have reported promising results in the use of highly hydrophilic linkers. This trend is expected to gain traction over more conventional approaches due to the recent approval of sacituzumab govitecan, an ADC that incorporates a CL2A acid-cleavable linker using PEG chains.
PEGylation has been shown to significantly increase the half-life of proteins in circulation. Its recent application to ADCs and the promising reports suggest that it might become one of the preferred methods for the development of novel ADC biopharmaceuticals.
ADCs are a unique class of biopharmaceuticals and many experts believe we are only just starting to realize their potential for multiple applications. Up until recently, linker chemistry had been a limiting step in the development of safe ADC therapies. Many of the early linkers showed poor stability in circulation leading to premature release and systemic toxicity.
But the discovery of highly stable, selective, and hydrophilic linkers has been quickly improving the biophysical properties of these conjugates. The recent boost in approvals and recent breakthroughs in linker chemistry indicate that many ADCs generated with alternative chemistries may soon reach the market.
You could also be interested in:
Join our email list to receive exclusive content featuring the most interesting industry and research news, biologics development tips pieced together by experts, res, company news, and exclusive limited-offers. Join a community of 80,000 subscribers and save up to 30% on your first order.