

Cart (0 Items)
Your cart is currently empty.
View ProductsIt looks like you are visiting from outside the EU. Switch to the US version to see local pricing in USD and local shipping.
Switch to US ($)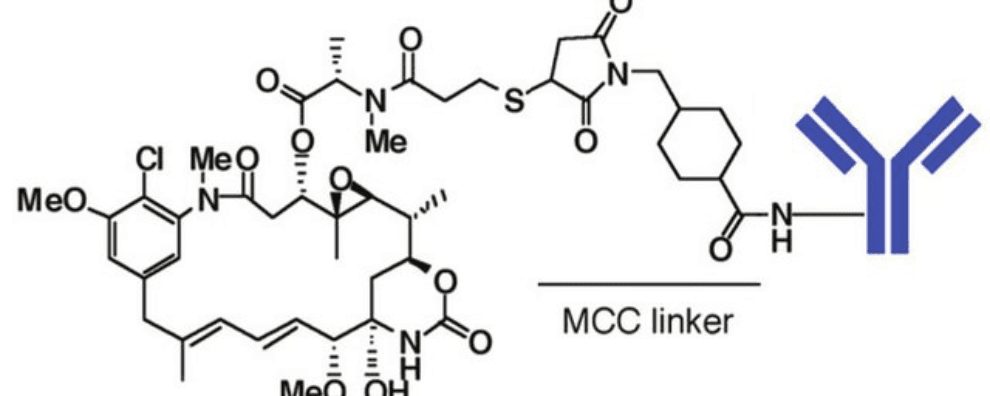 Antibody-drug conjugates
Antibody-drug conjugates
 Thomas Meyer
Thomas Meyer
Because linker design affects conjugation site, DAR distribution, and payload release, it is often developed together with broader antibody engineering strategies. ADC linkers can be divided into two major classes: cleavable and non-cleavable linkers. These ADC linker classes differ in how the payload is released, how stable the conjugate remains in circulation, and which therapeutic profiles they can support.
Linkers are on the interface of chemistry and biology in Antibody-Drug Conjugates (ADC). A linker can be cleaved from its payload and antibody carrier when exposed to specific chemical signals or enzymes, or it can be designed to integrate the payload and be released alongside it upon antibody degradation. These two main classes of linker chemistry, cleavable and non-cleavable, have been vital for the development of safer ADC biotherapies with wider therapeutic windows.
The most commonly used linkers contain disulfides and dipeptide motifs with antibody and payload-binding domains. The attachment of a linker-payload complex to the carrier antibody is controlled by the chosen bioconjugation method (i.e., chemical versus enzymatic conjugation). Moreover, the structure of the antibody-binding domain in a linker is what defines the number and distribution of drugs on an ADC, also known as the drug-to-antibody ratio (DAR).
In contrast, the attachment of the linker to the payload can be controlled by the linker technology (i.e., cleavable versus non-cleavable). The type of attachment defines the nature of the active payload species, drug release rates, on and off-target toxicities, and it further affects the solubility, stability, and therapeutic potency of an ADC. When developing a linker, particularly a cleavable linker, it is vital to ensure a balance between the plasma stability this linker grants the ADC and the efficiency of the payload’s release mechanism.
For this reason, the development of linkers for ADCs needs to be carried out using a holistic approach that takes into account the bioconjugation method and the nature and toxicity of the payload.
Choosing the right ADC linker is only one part of a successful antibody-drug conjugate program. ProteoGenix supports antibody drug conjugate development service end to end, from antibody discovery service and engineering to conjugation, analytical antibody characterization service, and production. Our ADC platform delivers high-potency conjugates with up to 90% of antibodies conjugated with the selected DAR, backed by 20+ years of experience in ADC development and 5 ADCs in clinical trials. For upstream support, our antibody discovery services provide 4 weeks from antigen to antibody and proprietary libraries with an average diversity of 10^10 clones per library, while our protein production platform gives access to 5 expression systems for fast screening and scale-up.
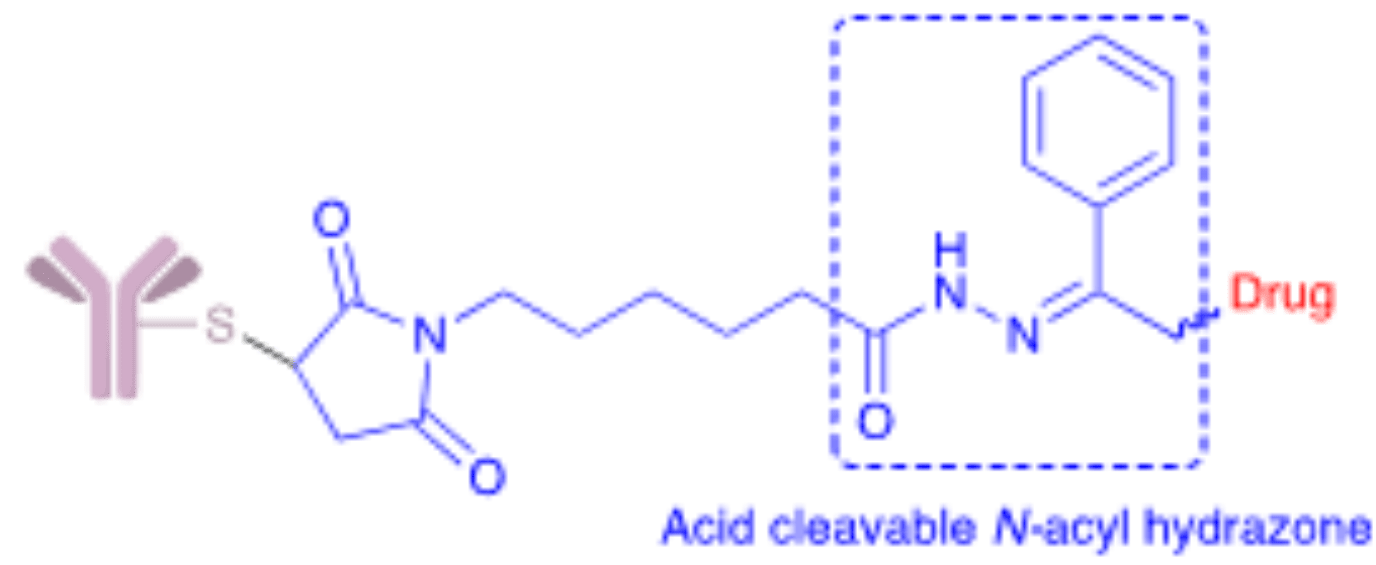
Hydrazone linkers are one of the earliest types of cleavable linkers used in ADC development. They are acid-labile molecules that remain relatively stable at physiological pH but release free drug upon hydrolysis in acidic lysosomes, late endosomes, or the extracellular acidic microenvironment of tumors.
Hydrazones have found limited success as linkers for ADC biopharmaceuticals carrying highly toxic cargoes. Gemtuzumab ozogamicin (Mylotarg®) remains as one of the classical examples of this technology first approved by the FDA in 2000, later withdrawn in 2010, and finally reapproved in 2017. This ADC contains the disulfide AcBut linker [4-(4-acetylphenoxy)butanoic acid] with acid-sensitive N-acyl hydrazone linkage. A combination that is also employed by inotuzumab ozogamicin (Besponsa®) approved in 2017.
The instability of hydrazone linkers is presumably caused by their slow hydrolysis under physiological conditions that leads to a progressive release of the drug in circulation and enhanced risk of systemic toxicity. For this reason, the use of hydrazone linkers is restricted to payloads with moderate toxicity. However, as ADCs move away from moderate to highly toxic cargoes, the use of these linkers has become more restricted.
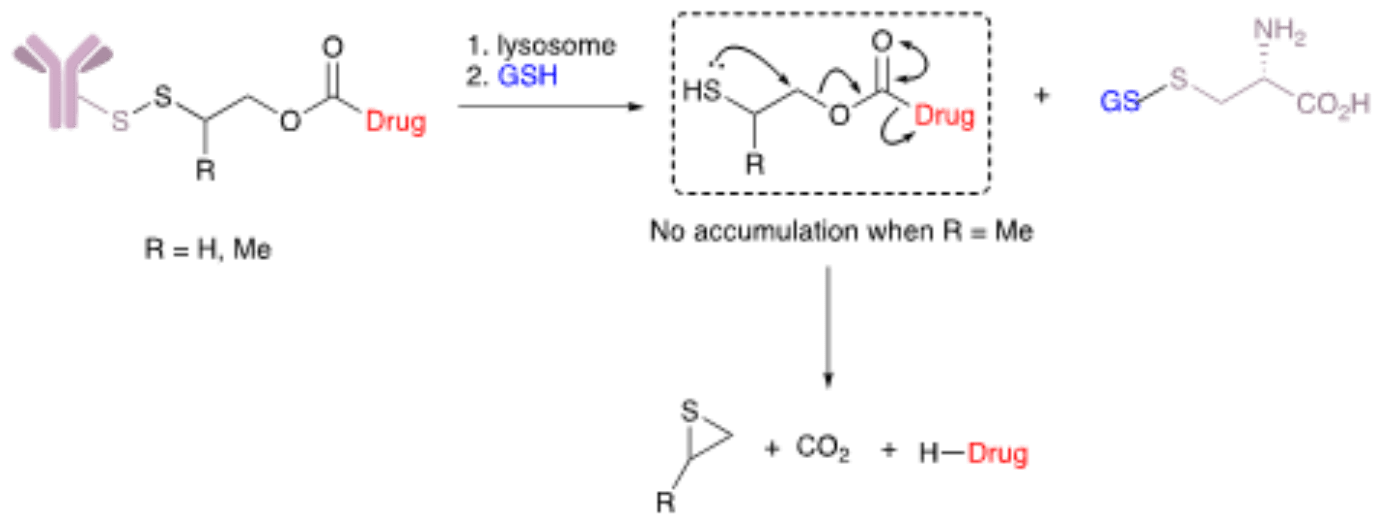
Reducible disulfide motifs belong to the most important class of cleavable linkers. They are found, alongside hydrazones, in the therapeutic ADCs Mylotarg® and Besponsa®. Disulfides are glutathione-sensitive linkers; thus, they cleave in the presence of high concentrations of the molecule which is heavily enriched in the cytosol (1-10 mM) and found at high concentrations in tumors’ microenvironments. To enhance the stability these linkers in circulation, methyl groups can be added close to the disulfide bond to provide steric protection.
This class of linkers has been successfully paired with maytansinoids with a thiol appendage, a class of potent cytotoxins. In these cases, a direct disulfide-bonding can be made between engineered cysteine residues in the antibody carrier and the thiols on maytansinoid payloads. Thus, the antibody carrier creates a shielding effect by protecting the disulfide bond from reduction without the need for modification and excessive steric protection. This strategy has been called linkerless chemistry because only a bond attaches the payload to its carrier.
The application of disulfide technology to a wider range of payloads has been made possible due to novel self-immolative carbamate linkers. These disulfate-carbamate linkers (e.g., β-mercaptoethyl-carbamate, −SCH(R)CH2OCO−) can be directly attached to cysteine residues on the antibody carrier and its in vivo stability is independent of the mechanism of payload release. This technology is considered traceless because it releases the payload in two steps: (i) release from the antibody carrier via disulfide cleavage of the thiol-cysteine adduct followed by (ii) immolation of the thiolate intermediate and subsequent release of the free unmodified payload. The immolation and subsequent efficiency of payload release can be further improved by substituting these linkers with various groups including methyl, ethyl, dimethyl, and cyclopropyl, among others.
Dipeptide linkers are one of the most successful classes of cleavable linkers used in ADC development. Since peptide linkers must be tuned for both plasma stability and efficient intracellular cleavage, they are often considered alongside antibody engineering services and conjugation constraints. They are present in the vast majority of ADCs currently in clinical trials including the FDA-approved brentuximab vedotin (Adcetris®) targeting human CD30. These linkers have been designed to target cathepsin B, a high-expression cysteine protease present in the lysosomal compartment of healthy individuals. The same enzyme has also been linked with tumor progression and reported to have high extracellular activity in necrotic tissues.
Dipeptide linkers typically result from a combination of hydrophilic high basicity residues susceptible to hydrolysis (e.g., Citrulline [Cit] and Arginine [Arg] are two of the most commonly used) with hydrophobic residues like Phenylalanine (Phe), Valine (Val), and Alanine (Ala), that guarantee dipeptide stability in circulation. Val-Cit and Val-Ala are two of the most successful dipeptide linkers developed to cleave in the presence of intracellular proteases.
Due to the steric bulk of the payload and antibody carrier, a spacer unit can be used to guarantee enzymes are able to access the peptide bond and cleave it quickly and effectively in vivo. Para-aminobenzyl carbamate (PABC) linkages have been used successfully for these purposes in Val-Cit-containing dipeptide linkers. PABC acts as a self-immolative spacer that, once the peptide bond is cleaved, undergoes spontaneous elimination releasing the naked and unmodified payload. Like disulfate-carbamate linkers, the use of PABC spacers ensures that linker cleavage remains independent from payload release. This property extends the applicability of this technology to a wider range of payloads while enhancing the stability of these constructs in circulation.
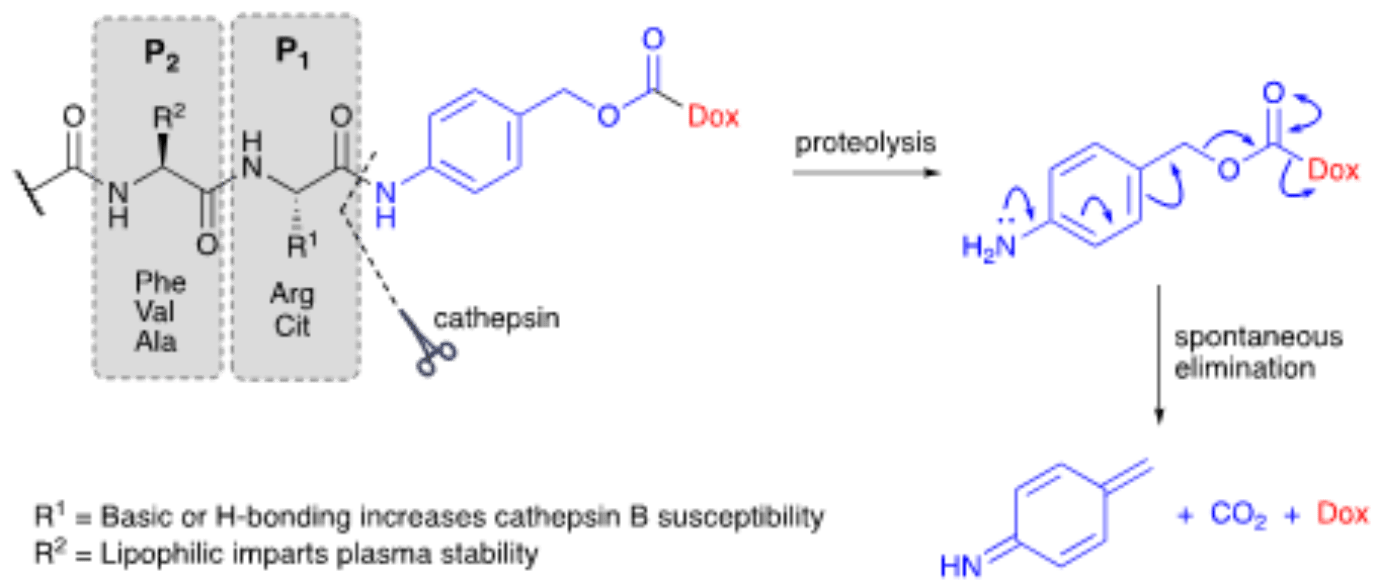
This type of linker has effectively replaced conventional hydrazone linkers as the predominant technology for ADC development. Of the many dipeptides tested in vivo, Val-Cit containing linkers showed the best plasma stability with some risk of premature release. This risk was shown to be highly dependent on the conjugation site as more solvent-exposed sites led to lower stability.
A strategy that has been successfully used to reduce this risk consists in the introduction of a third hydrophilic and acidic residue. The extension into a tripeptide configuration has proven to be beneficial for ADC stability without significantly affecting payload release rates. Several in vitro studies revealed that Glu-Val-Cit to be one the most stable and promising configurations. But more in vivo studies are necessary to further validate this methodology.
β-glucuronide linkers contain a hydrophilic β-glucuronic acid motif that targets β-glucuronidase, a hydrolytic lysosomal enzyme from the glycosidase class. Similar to cathepsins, these enzymes can be secreted by tumors to necrotic areas where they display restricted extracellular activity. For this reason, experts believe that ADCs with this type of linkage can act by internalizing and non-internalizing mechanisms.
The use of these linkers in conjugation with PABC spacers has proven to be stable in circulation with low levels of aggregation even at high DAR (i.e., DAR = 8). In experiments carried out using rat plasma, these linkers were shown to be more soluble and have longer half-lives in comparison to dipeptide linkers with comparable payload release efficiencies. The advantage of the β-glucuronic acid motif in comparison to Val-Cit-containing linkers appears to be its lower tendency for aggregation, which allows the development of ADCs with higher DAR and comparable pharmacokinetics (PK).
β-galactoside-containing linkers have recently emerged as a suitable alternative. These linkers target another lysosomal glycosidase – β-galactosidase. However, more in vivo studies are necessary to validate this approach for therapeutic ADC development.
Due to the known instability issues of dipeptide linkers, many new linkers have been developed targeting other intracellular enzymes. A few examples include motifs that target pyrophosphatases (e.g., pyrophosphate diesters motifs), acid phosphatases, and sulfatases. Although many of these linkers remain promising alternatives, more in vitro and in vivo studies are required to measure their plasma half-lives, payload release rates, and effect on the PK of the resulting ADCs.
Recently, a group of researchers at the University of Cambridge reported a dual-enzyme cleavable linker. The construct targets arylsulfatase A and β-galactosidase simultaneously and given its highly hydrophilic nature, it exhibits high selectivity and cytotoxicity. The researchers developed a linker containing a motif mimicking sulfatide – the natural substrate of arylsulfatase A – tied to a β-galactose motif. The resulting 3-O-sulfo-β-galactose linker displayed high lysosome-selectivity with cytotoxicities comparable to those observed in Val-Ala-PABC-containing ADCs.
The authors postulated that this kind of dual linker is an attractive alternative to more conventional linker chemistries. However, more studies are still necessary to demonstrate the efficiency of the technology for in vivo applications.
Thioether linkers are one of the most representative classes of non-cleavable linkers. Unlike all previously discussed linkers, thioethers never cleave from the payload, instead, their mechanism of action relies heavily on ADC internalization and lysosomal processing. Thioether linkers are generally more stable in circulation than all the previous classes of linkers, but they are also stricter in terms of the choice of antigens and payloads they can be conjugated with.
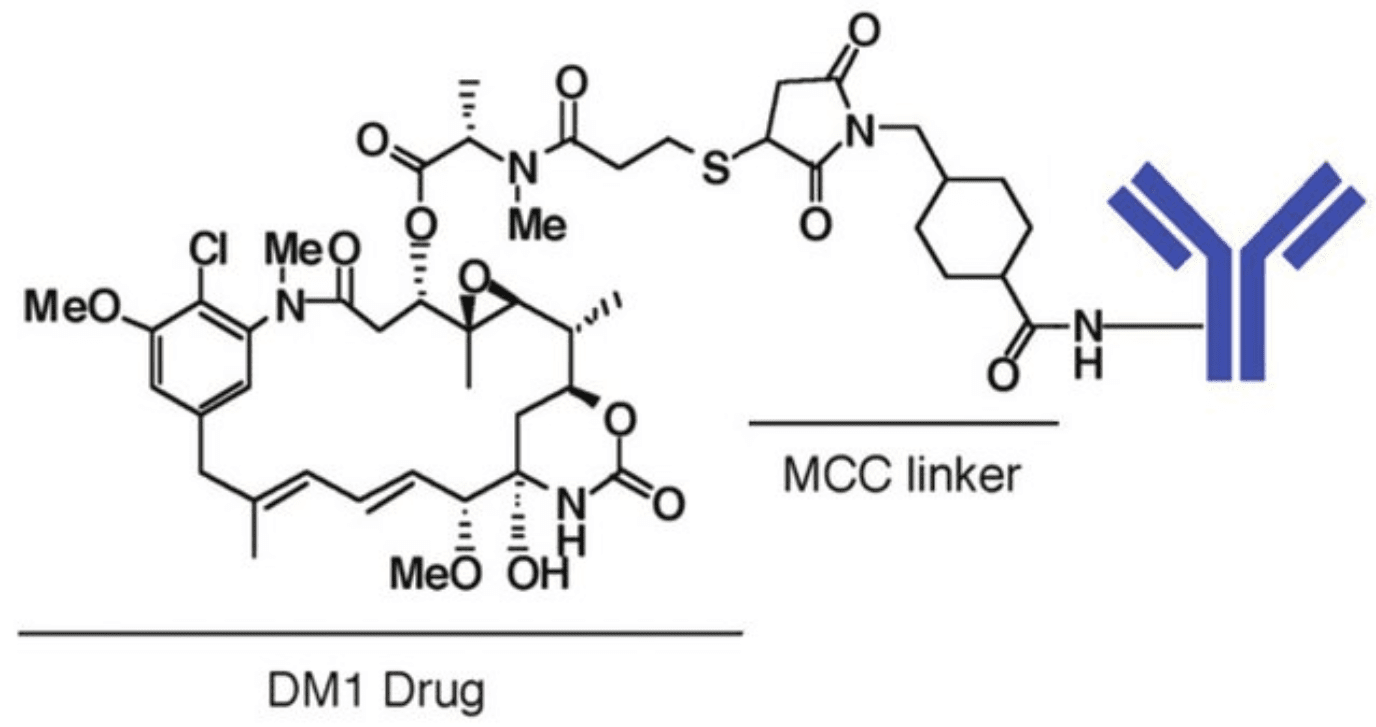
The most noticeable example of an ADC with a thioether-containing linker is trastuzumab emtansine (Kadcyla®). In this therapeutic ADC, the thioether linker contains cyclohexane and a maleimidomethyl group (maleimidomethyl cyclohexane-1-carboxylate, MCC linker) complexed with a thiol-containing maytansinoid payload (DM1).
Linker chemistry is constantly evolving as new strategies are devised and tested with the intent of maximizing the stability, safety, and therapeutic efficiency of ADCs. Currently, cleavable linker chemistries lead the market with dipeptide Val-Cit-containing linkers as one of the most widely used approaches used in ADC development.
The growing number of ADC approvals has recently revitalized the interest in the search for alternative linkers. Today, most alternative chemistries are based on more hydrophilic linkers such as the ones containing β-glucuronic acid motifs. Due to their hydrophilicity, these linkers are reported to reduce the risk of ADC aggregation and enhance the DAR with no detrimental effect on the PK of ADCs. More promising alternatives are expected to break into the market in the years to come.
You could also be interested in:
Join our email list to receive exclusive content featuring the most interesting industry and research news, biologics development tips pieced together by experts, res, company news, and exclusive limited-offers. Join a community of 80,000 subscribers and save up to 30% on your first order.