
Cart (0 Items)
Your cart is currently empty.
View ProductsIt looks like you are visiting from outside the EU. Switch to the US version to see local pricing in USD and local shipping.
Switch to US ($)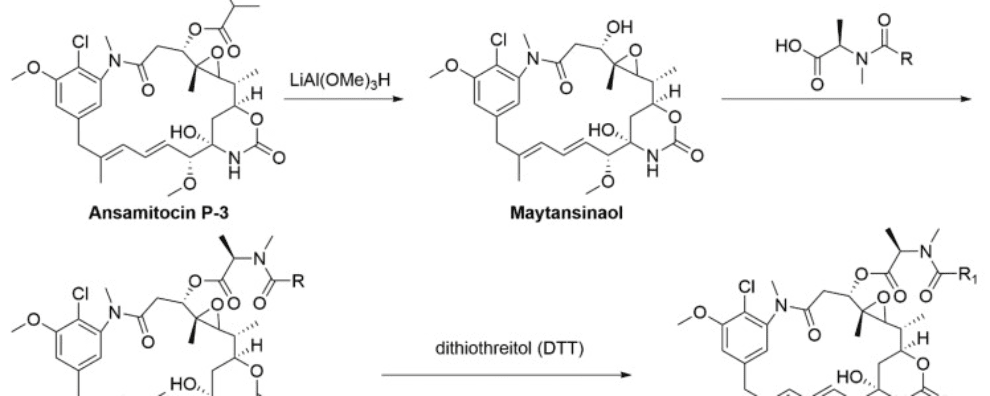 Antibody-drug conjugates
Antibody-drug conjugates
 Thomas Meyer
Thomas Meyer
Microtubules are highly conserved and ubiquitous structures at the center of many eukaryotic cellular processes including cell division, transport, and cell structure maintenance. Disrupting the assembly of α- and β-tubulin heterodimers – the main components of microtubules – arrests the cell cycle in the G2/M phase and eventually causes cell death by apoptosis. Microtubules are a major target for anticancer drug discovery due to the enhanced proliferation of cancer cells in comparison to healthy and nonproliferating cells.
Tubulin-targeting drugs are classified according to their mechanism of action and specific binding site:
Early tubulin inhibitors used in therapeutic applications were natural products or synthetic derivates such as taxanes (e.g., paclitaxel) and vinca alkaloids (e.g., vinblastine). However, many of these products resulted in low cytotoxicity and often led to the quick development of drug resistance.
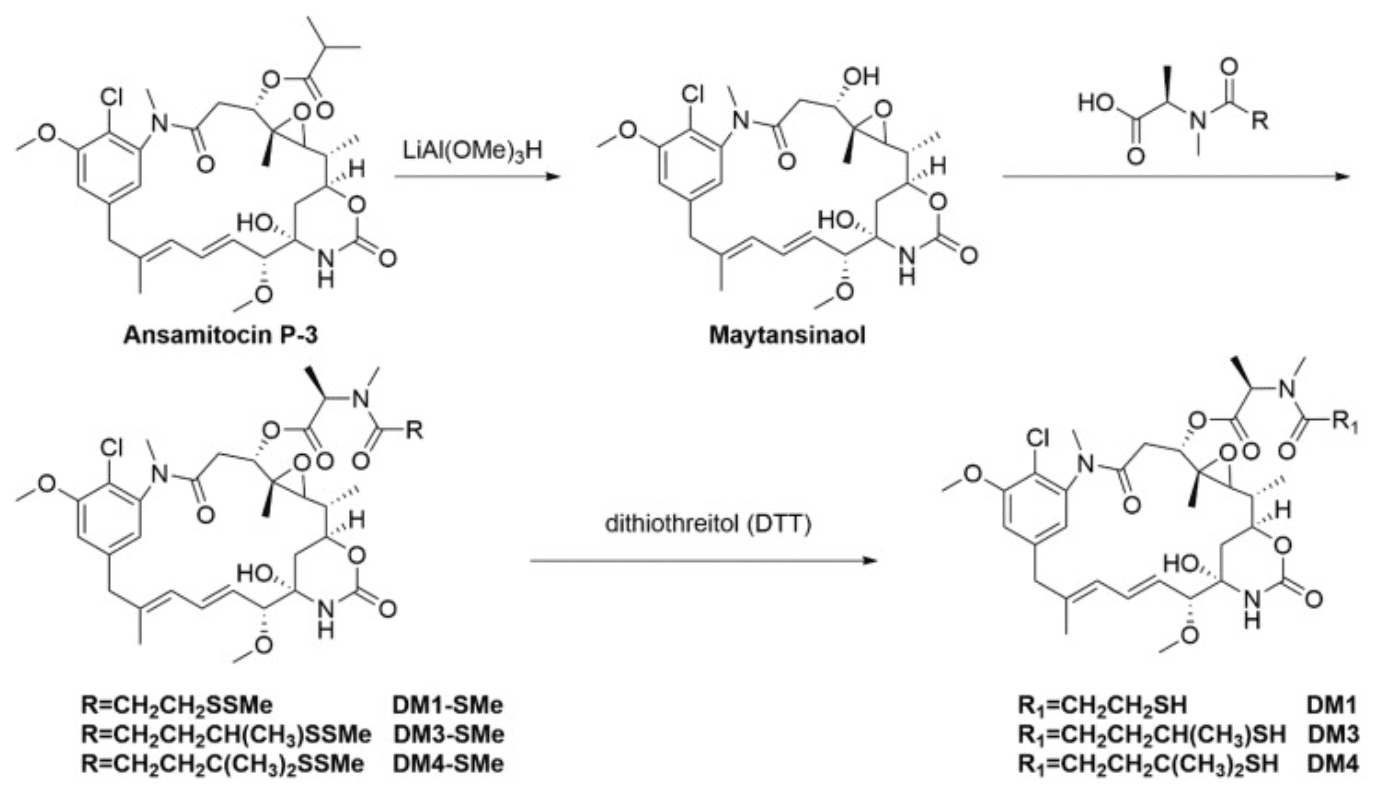
These limitations made them inadequate for ADC development. Newer generations of tubulin inhibitors achieved higher potencies. One of the first of the new generation was maytansine.
Maytansine was initially isolated from an alcoholic extract of the bark of African shrubs. The original compound was found to have 100- to 1000-fold higher potency than doxorubicin or paclitaxel, two early tubulin-targeting agents. As a monotherapy, maytansine was found to cause severe side effects and result in poor efficacy due to its lack of tumor specificity and narrow therapeutic window. But its enhanced toxicity made this drug and corresponding derivates highly desirable as payloads of ADCs.
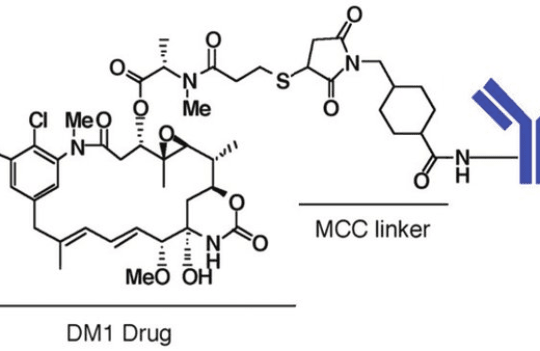
Maytansinoids are derivates of maytansine known to bind to the maytansine binding site and destabilize the microtubule assembly dynamics. These structurally complex drugs are typically synthesized from ansamitocins, natural compounds sourced from the fermentation of Actinosynnema pretiosum, an actinobacterium. Derivates DM1 (N2′-deacetyl-N2′-(3-mercapto-1-oxopropyl)-maytansine) and DM4 (N2′-deacetyl-N2′-(4-mercapto-4-methyl-1-oxopentyl)-maytansine) bear thiol groups, allowing covalent binding via cleavable disulfide bonds to monoclonal antibodies. The process of semi-synthesis of these drugs (starting from a natural biological compound) is the preferred approach at a large scale because due to the structural complexity of these drugs, de novo synthesis has a prohibitive cost.
The first maytansinoid-based ADC to reach the clinic was trastuzumab emtansine (Kadcyla, DM1-based ADC). Approved by the FDA in 2013, this ADC is currently used to treat HER2-positive metastatic breast cancer.
Auristatins are derived from dolastatin-10, a natural product isolated from the sea hare Dolabella Auricularia from the Indian Ocean and the coastal waters of Japan. This compound and its analogs bind at the vinca alkaloid binding site on α-tubulin, blocking its polymerization, causing metaphase arrest, and leading to cell death by apoptosis. The vast majority of ADCs approved by the FDA bear auristatins as payloads including Adcetris® (brentuximab vedotin, approved in 2011), Polivy® (Polatuzumab vedotin, approved in 2019), and Padcev® (enfortumab vedotin, approved in 2019), all bearing a valine-citrulline protease cleavable linker. A dozen more ADCs of this class are currently in the clinical pipeline.
As monotherapies, auristatins showed no significant therapeutic efficacy at the maximum dose tolerated. However, this class of drugs has thrived as payloads of ADCs. The most common payloads of this class used in ADC development are fully synthetic and include monomethyl auristatin E (MMAE) and monomethyl auristatin F (MMAF), estimated to have lower toxicity than MMAE and enhanced aqueous solubility. Due to being fully synthetic, these drugs have an advantage over maytansinoids due to the ease and cost-efficient synthesis process. Additionally, it is also easier to modify their structure to optimize their therapeutic properties.
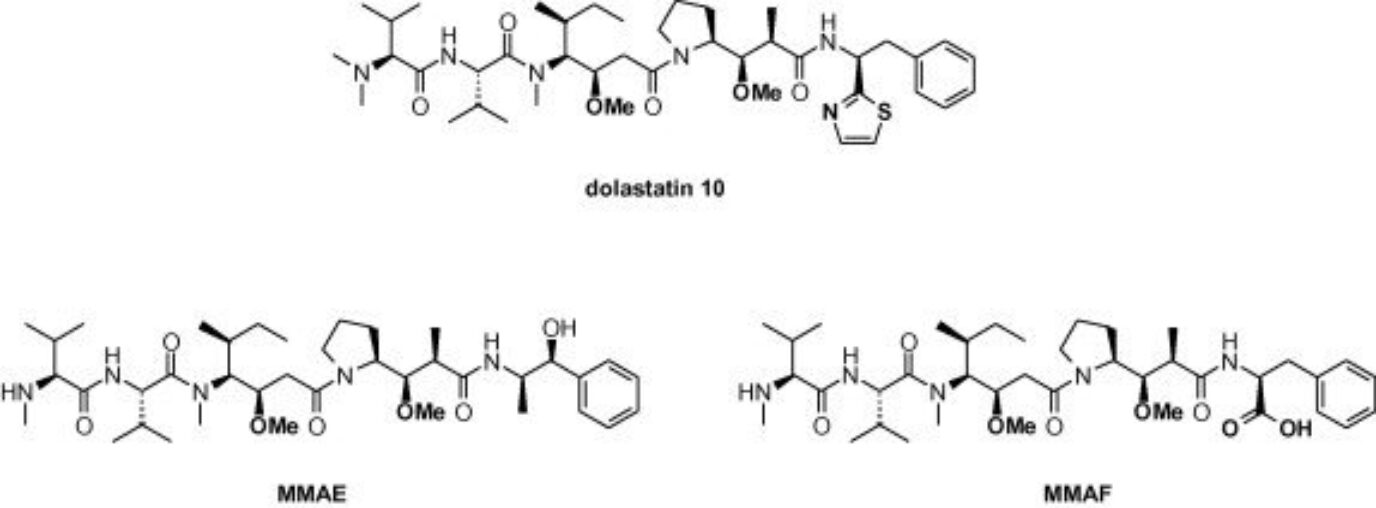
Due to the ease of synthesis, this class of drugs has continued to evolve in recent years. Most modifications on the auristatin molecule have been focused on the N- or C-terminus, without alterations to the core peptides. These modifications have been made with the intent of increasing the drug’s hydrophilicity since hydrophobic drugs make poor ADC payloads resulting in protein aggregation and fast clearance.
Although maytansinoids and auristatins remain the dominant payloads of ADCs, other drugs with similar mechanisms of action are continuously being developed. These emerging classes include cryptophycins, tubulysins, and hemiasterlins.
Cryptophycins are a class of 16-membered cyclic depsipeptides, first isolated from terrestrial blue-green algae. These drugs have proven to be potent antimitotic agents but preclinical trials have yet to demonstrate their in vivo efficacy in conjugation with antibodies. One of the main disadvantages of this class of drugs is their structural complexity, making them harder to synthesize.
Tubulysins are natural products isolated from myxobacterial cultures. Structurally, tubulysins are tetrapeptides containing D-methylpipecolate (D-Mep), L-isoleucine (L-Ile), L-tubuvaline (L-Tuv), and L-tubuphenylalanine (L-Tup) residues. Tubulysin D is one of the most potent cytotoxic agents within this class with IC50 values ranging from 0.01 and 10 nM. These compounds act similarly to maytansinoids by arresting the cell cycle between the G2 and M phase leading to cell death by apoptosis. Several hydrophilic analogs of tubulysins are currently under development.
Hemiasterlin is another natural product isolated from marine sponges. The parental compound and its synthetic analogs are tripeptides that bind to the vinca binding site, inhibiting polymerization, triggering mitotic arrest, and subsequent apoptosis. Full de novo synthesis has been achieved for these compounds, making them very promising for ADC development.
Join our email list to receive exclusive content featuring the most interesting industry and research news, biologics development tips pieced together by experts, res, company news, and exclusive limited-offers. Join a community of 80,000 subscribers and save up to 30% on your first order.