
Cart (0 Items)
Your cart is currently empty.
View ProductsIt looks like you are visiting from outside the EU. Switch to the US version to see local pricing in USD and local shipping.
Switch to US ($)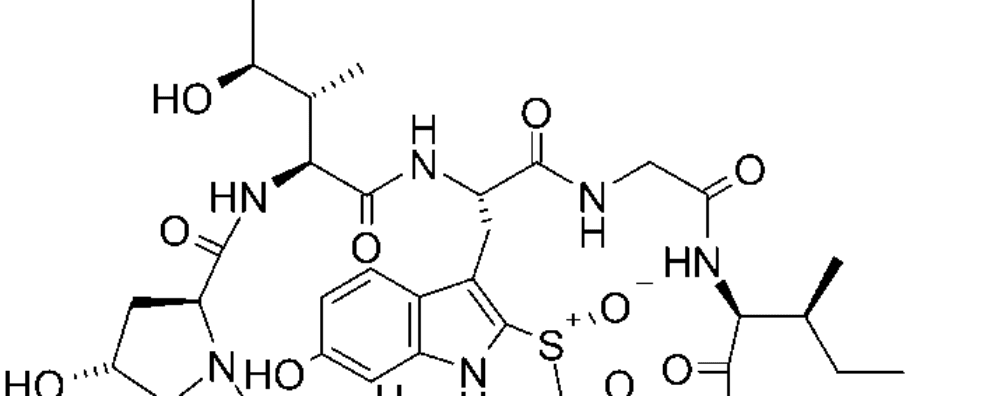 Antibody-drug conjugates
Antibody-drug conjugates
 Thomas Meyer
Thomas Meyer
The vast majority of antibody-drug conjugates (ADCs) in the clinic use tubulin-targeting agents as payloads (auristatins and maytansinoids). These compounds are highly effective at targeting proliferative cells by arresting the mitotic cycle and leading to cell death via apoptosis. However, as these emerging therapeutics attempt to tackle solid tumors, other classes of payloads have come into play.
Compounds that interfere with the structural integrity of DNA or impact the transcription process have a tremendous value as anticancer agents. They act independently of the growth phase of the cell and exhibit higher potency than tubulin inhibitors, making them desirable as payloads for ADC development.
The major families of DNA-damaging agents include:
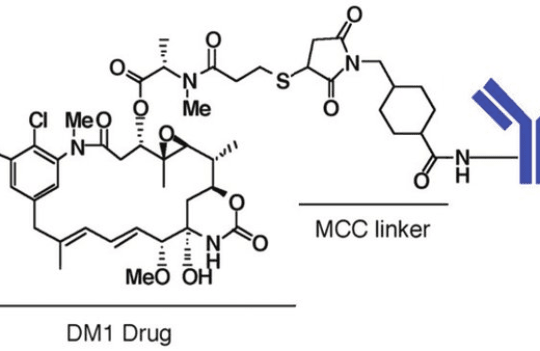
Most of these families of drugs can be conjugated to monoclonal antibodies via dipeptide linkers such as valine-citrulline or valine-alanine. These linkers are designed to be cleaved by lysosomal proteases (cathepsin B) and remain one of the most successful chemistries used for ADC applications.
Up to recently, the majority of ADCs carrying DNA-damaging agents in the clinic used calicheamicin as a payload such as gemtuzumab ozogamicin (Mylotarg®, 2017) and inotuzumab ozogamicin (Besponsa®, 2017). But the use of alternative families of DNA-damaging drugs seems to be gaining ground in recent years due to the development of safer linker chemistries.
The proof is the approval of trastuzumab deruxtecan (Enhertu®) in 2019, an ADC conjugated to DXd, a potent DNA topoisomerase inhibitor and an exatecan analog of the camptothecin family. In 2020, another ADC coupled with a topoisomerase inhibitor was approved – sacituzumab govitecan (Trodelvy®). The new drug was conjugated to an SN-38 payload, an irinotecan analog derived from camptothecin. In 2021, ADC Therapeutics was granted marketing approval for loncastuximab tesirine-lpyl (Zynlonta®), a potent ADC carrying SG3199, a pyrrolobenzodiazepine (PBD) dimer, and an alkylating agent. Other ADCs conjugated to similar drugs are currently in clinical trials.
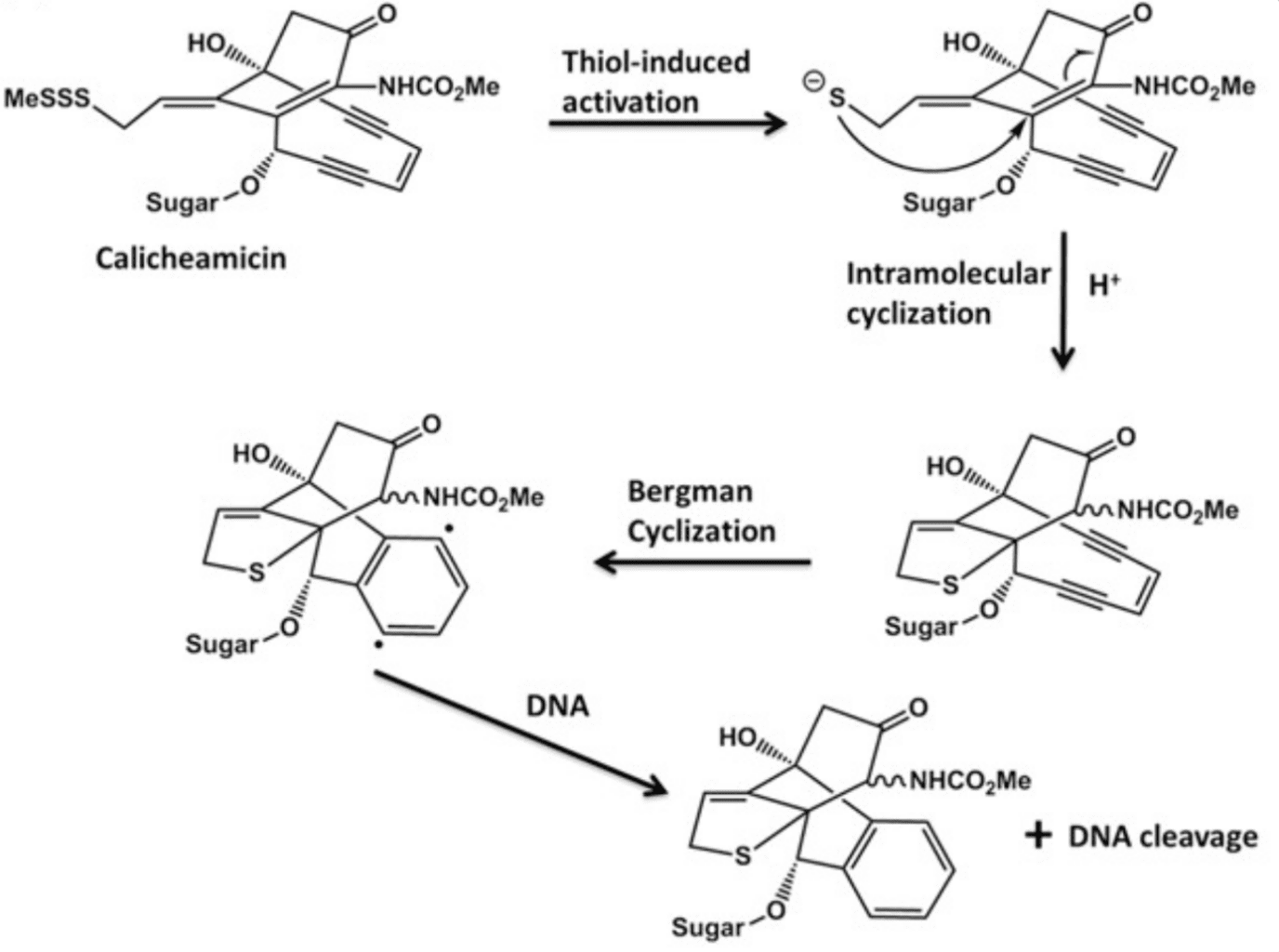
Calicheamicin was initially isolated from Micromonospora echinospora, an actinomycete found in soil. Calicheamicin γ1 is the most widely used and the most potent within this family of small drugs. These compounds lead to DNA cleavage after undergoing thiol-induced activation in the presence of glutathione, heavily enriched in the intracellular environment. In turn, the activation results in a series of molecular rearrangements (Bergman cyclization) that culminate in the production of diradicals that cleave the double strands of DNA, resulting in cell death.
Calicheamicin and its derivates are non-specific attacking indiscriminately the DNA of both cancer and healthy cells. As a result, they cannot be used as monotherapies but serve as highly desirable warheads for ADCs.
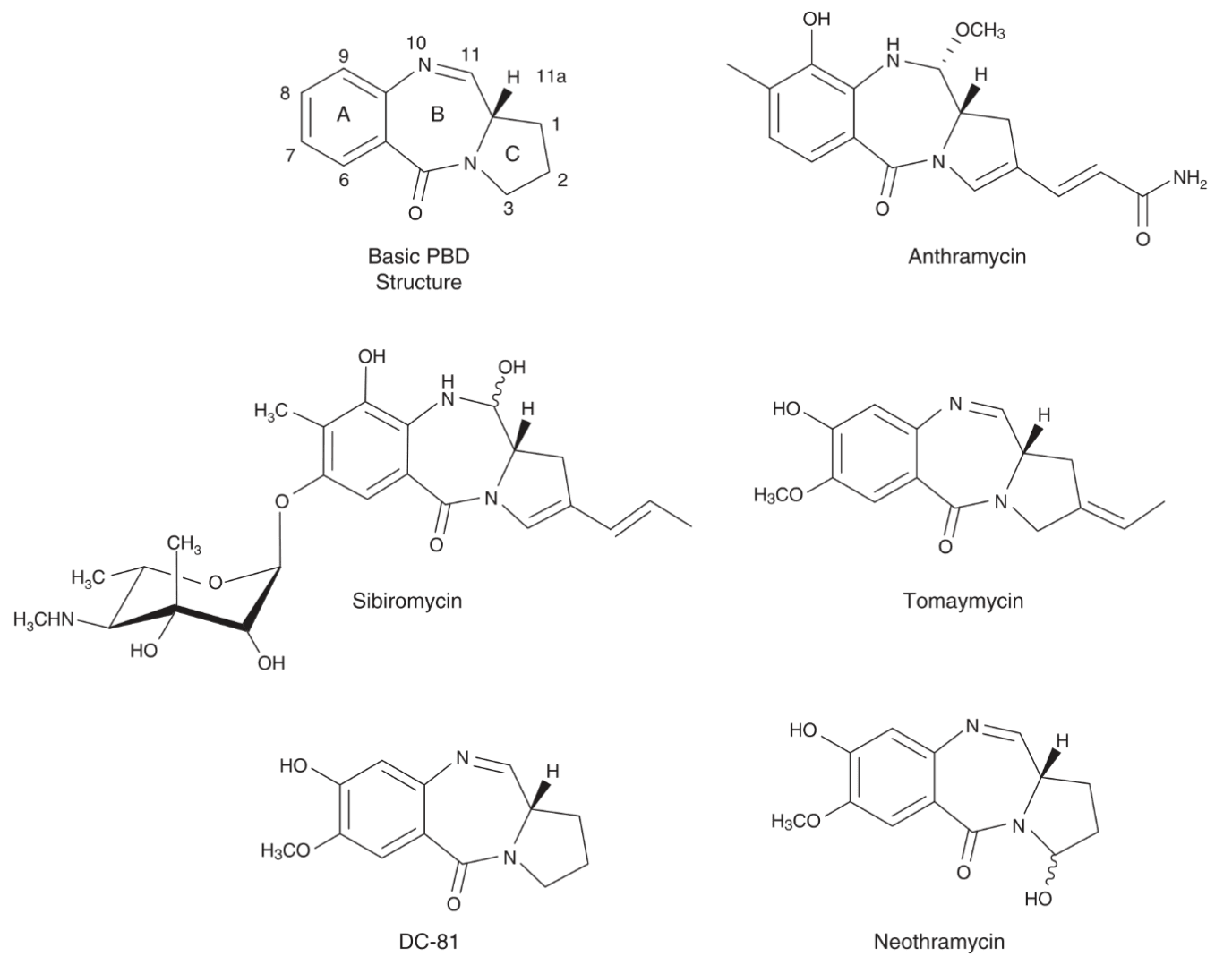
PBD monomers were first isolated from Streptomyces bacteria. They are all tricyclic molecules consisting of an aromatic A-ring, a 1-4-diazepin-5-one B-ring, and a pyrrolidine C-ring. PBD dimers emerged shortly after when researchers discovered that the conjugation of two PBD monomers via a propyldioxy linker rendered compounds 600x more potent. Since then, this class has become an invaluable source of anti-tumor and antibiotic agents.
These compounds act as DNA minor groove binding agents, cross-linking specific DNA segments. By doing so, these drugs hinder cell division and lead to cell death. These dimers are non-specific and show high activity in the picomolar range. This makes them unsuitable as chemotherapeutic agents, but highly valuable for ADC development. Despite being a natural product, these drugs can be produced synthetically which greatly streamlines their production process.
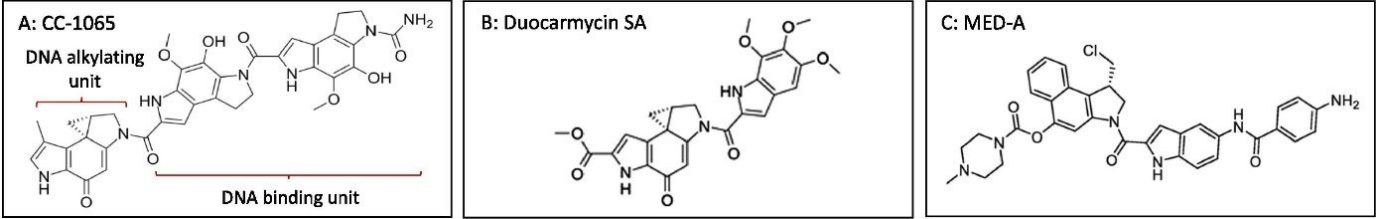
Like other DNA-damaging agents, duocarmycins have a natural origin. They were first isolated from Streptomyces zelensis in the late 1970s and found to contain a DNA-binding region and a DNA-alkylating region, possessing a spirocyclic cyclopropapyrroloindole (CPI) moiety. Many synthetic derivates of the first duocarmycin (CC-1065) ended up maintaining these structural features.
With IC50 values raging from picomolar to nanomolar levels, duocarmycins have captured the attention of the scientific community due to their immense anticancer activity. These drugs bind the minor groove of DNA molecules at AT-rich regions and alkylate adenine residues at the N3 position. The process results in cleavage of the DNA strand and subsequent cell death. Duocarmycins act on cells independently of their growth stage (proliferating and non-proliferating cells). This means that cancer cells showing hypoxic, chemoresistant, and stemness phenotypes are quite sensitive to duocarmycins, making them extremely useful to treat patients at the later stages of malignancy. These drugs are also better than tubulin-inhibitors at targeting solid tumors.
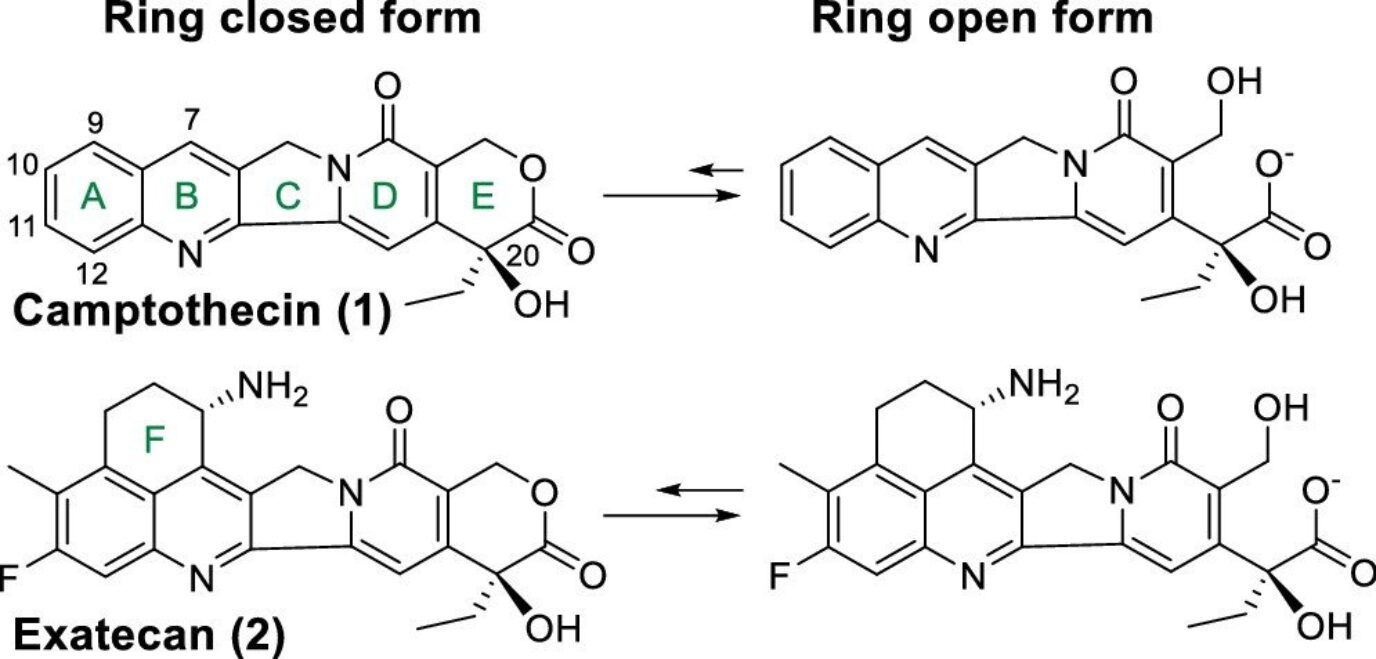
Camptothecin analogs are quickly gaining importance as payloads for ADC development as seen by the recent approvals of trastuzumab deruxtecan (Enhertu®) and sacituzumab govitecan (Trodelvy®) in 2019 and 2020, respectively. Camptothecin was first isolated from the Chinese ornamental tree Camptothecaacuminata in the 1980s. Unlike the previously described DNA-damaging agents, this drug acts as a potent DNA topoisomerase I inhibitor. This key enzyme is responsible for cleaving the double-stranded DNA, partially unwinding it, and reannealing the strands to relieve tension. By binding this enzyme, camptothecin and its analogs prevent reannealing leading to the accumulation of partially cleaved DNA and subsequent cell death.
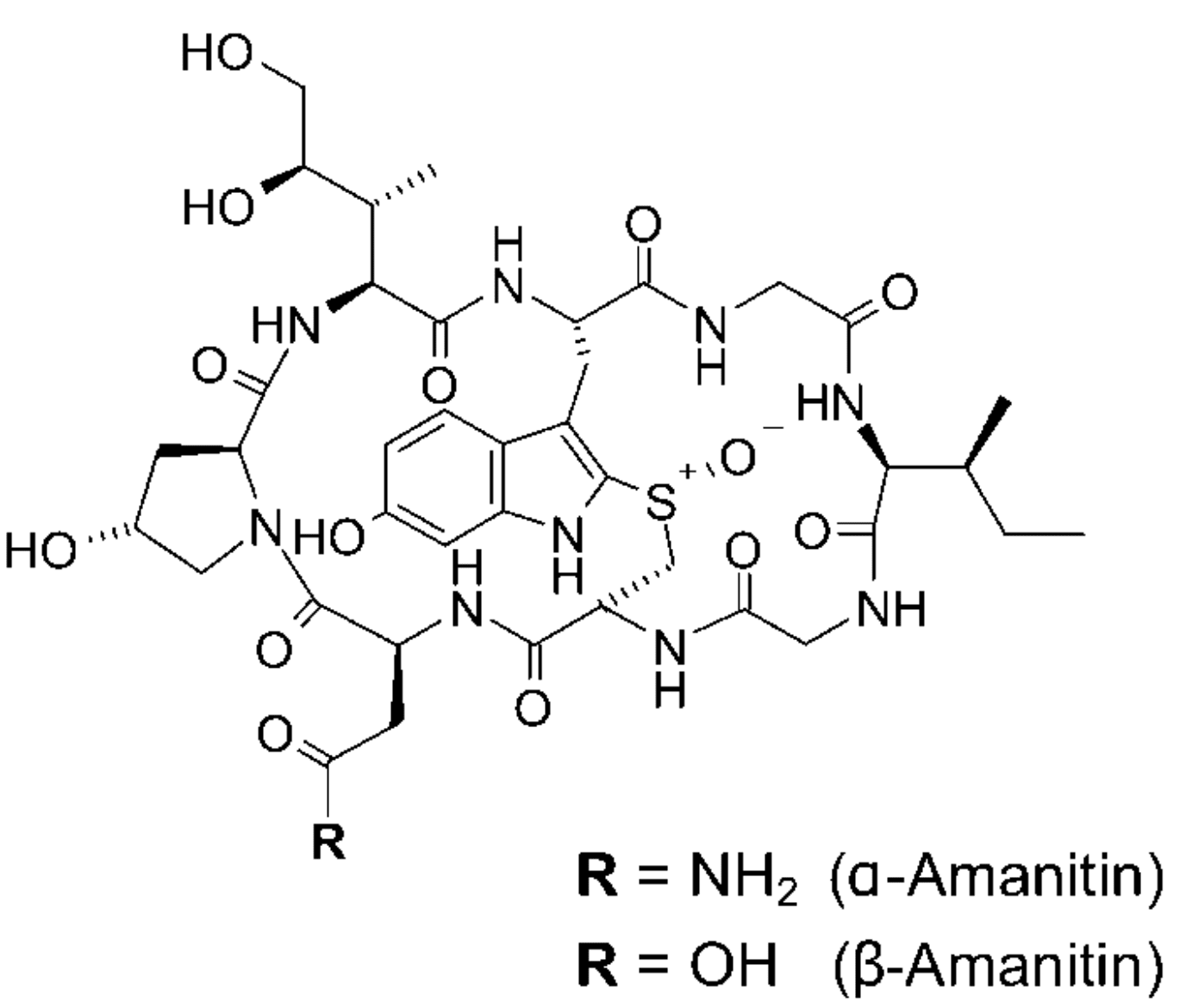
Amanitin-based ADCs represent a new class of biotherapeutics with a novel mode of action. Unlike the previous drugs, this payload inhibits RNA polymerase II thus affecting the process of transcription. Collectively, this class of drugs is known as amatoxins, they consist of bicyclic octapeptides occurring naturally in basidiomycetes mushrooms of the genus Amanita. Amatoxins are well-known due to the decades of study of their toxic effects after mushroom ingestion, responsible for 95% of all fatal mushroom intoxications around the world.
These toxins are extremely resistant to heat, proteolytic cleavage, and acid degradation. Moreover, they are extremely water-soluble, making them optimal for ADC applications. They bind strongly to eukaryotic RNA polymerase II leading to a 1000-fold decrease in transcription and protein synthesis. Amanitin affects not only proliferative cells but also resting cells, unlike tubulin inhibitors and some DNA-damaging agents.
HDP-101 is one of the most advanced amanitin-based ADCs in the clinical pipeline. This ADC is conjugated via a protease cleavable linker and it targets the B-cell maturation antigen, a highly specific marker for malignant plasma cells. HDP-101 is expected to be one of the first of its type to obtain marketing approval by the FDA.
Improved linker chemistry has allowed the use of increasingly potent payloads. Experts argue that the next generation of these biotherapeutics will evolve to target even more specific and less abundant disease markers which will, in turn, allow the diversification of the mechanisms of action of ADCs and the widening of their therapeutic window.
This precise targeting requires two achievements: (i) the development of payloads active in the picomolar-femtomolar range and (ii) the improvement of antibody internalization rates. The latter translates into the use of alternative biotherapeutics such as bispecific antibodies, which have shown enhanced internalization and further allow the combination of the ADC mechanism of action with an immunotherapeutic response – yet to be achieved.
Another need that will drive innovation in payload discovery is the need to handle drug resistance. Many cancer cells, particularly those in solid tumors, are recalcitrant to the action of common cytotoxic drugs or quickly develop resistance mechanisms against them. Some experts believe that DNA damaging agents can overcome these issues, making them one of the preferred payloads of next-generation ADCs.
Join our email list to receive exclusive content featuring the most interesting industry and research news, biologics development tips pieced together by experts, res, company news, and exclusive limited-offers. Join a community of 80,000 subscribers and save up to 30% on your first order.