
Cart (0 Items)
Your cart is currently empty.
View ProductsIt looks like you are visiting from outside the EU. Switch to the US version to see local pricing in USD and local shipping.
Switch to US ($) Antibody-drug conjugates
Antibody-drug conjugates
 Thomas Meyer
Thomas Meyer
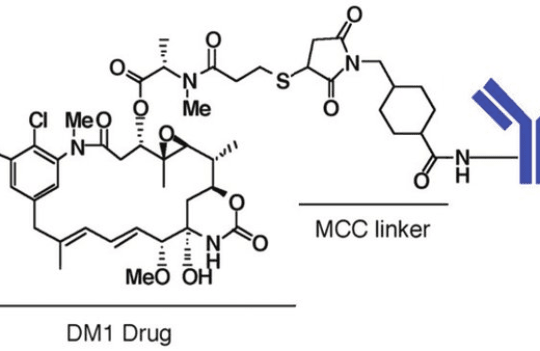
Linker technology allows the conjugation of a monoclonal antibody to a specific payload. Linkers dictate an ADC’s stability in plasma, its mechanism of action (internalized or non-internalized), therapeutic efficiency (PK/PD), and therapeutic window. Over the years, different methods for tethering a payload to its carrier have been developed. These can be broadly classified as cleavable or non-cleavable.
Non-cleavable linkers are stably linked to the payload and the pair is only released from the carrier upon endocytosis and lysosomal processing to release the cytotoxic cargo attached to its linker. Both offer different advantages and limitations. In contrast, cleavable linkers represent the major class of ADC linkers. As their name indicates, these small molecules cleave in response to specific chemical signals. In this way, they either require internalization and lysosomal processing or just release a membrane-permeable payload in the proximity of a tumor, killing both the antigen-positive and antigen-negative cells – called the “bystander effect.”
On the one hand, non-cleavable linkers are considered more stable with lower off-target toxicity and longer plasma half-lives. However, the stable crosslink may hinder the activity of some drugs because the linker is prone to altering the structure of its conjugate.
On the other hand, cleavable linkers are cleaved easily in response to environmental differences such as redox potential, enzymes, and pH, among others. In this case, since cleavable linkers release the payload, it is easier to estimate the cytotoxic potential of the drug. In other words, cleavable linkers are compatible with a wider range of drugs because they do not modify their structure nor their native activity.
Moreover, cleavable linkers allow the development of non-internalizing ADCs. This emerging subclass of ADCs forgoes the need for endocytosis to exert a therapeutic effect. Thus, they bypass the risk of inefficient intracellular trafficking and consequently reduced the therapeutic effect. Moreover, monoclonal antibodies used for these applications can be designed to target non-internalizing and low-expression antigens. The most important limitation of this approach is the high risk of generating off-target toxicity.
The development of new cleavable linkers is an active area of research. The linker’s main functions are to ensure a high selectivity and potency of ADCs, for this reason, they are optimized according to three key properties: (i) high stability in circulation; (ii) high solubility to avoid the formation of inactive aggregates; and (iii) efficient release of the payload. A cleavable linker comprises two parts: the antibody-binding domain and the payload-binding domain. The first domain dictates ADC stability and homogeneity, while the second domain dictates the mechanism of action and the efficiency of the payload release.
Interestingly, antibody engineering can be used to reduce the off-target toxicity of cleavable ADCs. For instance, bioconjugation sites in the antibody carrier can be attached to less solvent-accessible areas. In this way, the linker becomes less exposed to chemical and enzymatic triggers, therefore increasing its stability and slowing release rates.
At the moment, these linkers can be classified into two subclasses.
Chemically labile linkers can be further classified as acid cleavable or reducible linkers. Both are clinically established methods as several FDA and EMA-approved ADCs employ this linker chemistry, moreover, much more chemically labile ADCs are currently undergoing clinical trials.
Acid-cleavable linkers
This type of linker chemistry exploits the natural acidity of endosomes and lysosomes (pH 4.5-6.2) which contrasts with the neutral pH (7.4) of the plasma. Mylotarg® and Besponsa® are two of the ADCs currently on market to have been developed with this type of chemistry. The most commonly used acid-cleavable linkers are hydrazones which have consistently proven to be stable at neutral pH and labile at pH 4.5. Other acid labile groups have been used for this approach including the combination of carbonate linkers with alcohol-containing payloads.
This approach is most useful when designing ADCs with conventional mechanisms of action (internalizing ADCs); however, acid-cleavable linkers able to release payloads extracellularly have been previously reported. Despite the early successes of this type of chemistry, acid-labile linkers are only rarely used today because they remain limited to moderately toxic payloads.
Reducible or disulfide linkers
Disulfides are the most prominent class of chemically labile linkers. This linker chemistry is stable at physiological pH but susceptible to nucleophilic attacks from thiols. In the plasma, only human serum albumin (HSA) contains thiols but their reactivity is limited due to the reduced solvent exposure of these groups. In contrast, the cytosol is enriched in glutathione (GSH), with exposed and highly reactive thiol groups. Its presence offers the opportunity to develop disulfide linkers for highly selective intracellular release. Moreover, elevated GSH levels are often a feature of oxidative stress associated with tumors adding another layer of selectivity to this type of chemistry.
Several strategies have been used to increase the efficiency of these linkers, namely steric substitution which, at an intermediate level, was shown to increase ADC plasma stability while maintaining a good intracellular cleavage efficiency.
Based on this chemistry, researchers at Genentech have developed a linkerless technology where disulfide-bonding is formed between engineered cysteine residues in the antibody carrier and thiols in maytansinoid, a cytotoxic agent acting as a microtubulin polymerization inhibitor. This technology has demonstrated enhanced linker stability and increased antitumor activity.
As their name indicates, this type of linker chemistry makes use of intracellular or extracellular enzymes for payload release. A relatively common enzyme exploited in payload release is cathepsin B, a cysteine protease restricted to the lysosomal compartment in healthy cells and overexpressed in the extracellular environment in many cancers. Cathepsin B cleaves the peptide bond between dipeptide linkers containing valine (Val) and citrulline (Cit) and p-aminobenzylalcohol (PAB).
Valine-citrulline is the most commonly used dipeptide linker in current enzyme-dependent linker chemistry. Initial constructs using this dipeptide linker showed instability in plasma; however, further studies revealed this instability to be caused by the premature degradation of the linker by unspecific proteases. Further modifications in the small peptide allowed reducing the exposure of the cleavable sites and enhance the stability of this enzyme-cleavable linker.
Glycosidase-cleavable linkers are another commonly used enzyme exploited in ADC development. These hydrolytic enzymes are typically confined to the lysosomal compartment, but like cathepsin B, they can be secreted by tumor cells in necrotic areas. β-glucuronidase and β-galactosidase cleavable linkers are the most used type of glycosidase-cleavable chemistry currently in use. The first is the more commonly used of the two and it catalyzes the breakdown of β-glucuronic acid residues into polysaccharides at lysosomal pH.
Cleavable linkers offer a greater degree of control over drug release mechanisms. This type of chemistry is also more flexible than its non-cleavable counterpart, allowing diversified mechanisms of action (internalizing and non-internalizing ADCs), targeting a wider range of antigens, and conjugation with drugs with moderate to high toxicity.
Cleavable chemistry thus remains the dominant type of chemistry for most applications. The development of the linker must be done in parallel with the design of the antibody and the selection of conjugation sites and strategy. Experts agree that, when it comes to linker chemistry, there isn’t a one-size-fits-all solution. On the contrary, the ideal linker will depend on the nature of the tumor (i.e., solid or hematological), progression of the disease, and type of cancer.
As a general rule, linkers can be optimized by achieving a balance between the stability of the ADC in circulation (i.e., making linkers less exposed to solvents) and the efficiency of the payload release (i.e., fast cleavable in response to specific chemical signals).
You could also be interested in:
Join our email list to receive exclusive content featuring the most interesting industry and research news, biologics development tips pieced together by experts, res, company news, and exclusive limited-offers. Join a community of 80,000 subscribers and save up to 30% on your first order.