
Cart (0 Items)
Your cart is currently empty.
View ProductsIt looks like you are visiting from outside the EU. Switch to the US version to see local pricing in USD and local shipping.
Switch to US ($) Antibody-drug conjugates
Antibody-drug conjugates
 Thomas Meyer
Thomas Meyer
Monoclonal antibodies in ADCs are designed to deliver payloads to specific targets. The specificity and high antigen-affinity of these glycoproteins make them the ideal partners of cytotoxic drugs with proven in vitro therapeutic efficiency, but high in vivo toxicity.
Partnering these two components for optimal efficiency is an active area of research and several methods are recurrently used both at the small and large scale. However, one of the challenges in antibody conjugation is controlling the drug load in the ADC molecule.
Many of the early methods of conjugation resulted in heterogeneous mixtures of ADC with variable drug loads. This variability has for long hindered the development and use of ADCs for therapeutic applications because high loads lead to aggregation, lower tolerance, and faster clearance, while low loads result in suboptimal efficacy. For this reason, the precise control of antibody conjugation sites is vital to ensure optimal ADC pharmacokinetics (PK), pharmacodynamics (PD), and general efficiency for non-therapeutic applications.
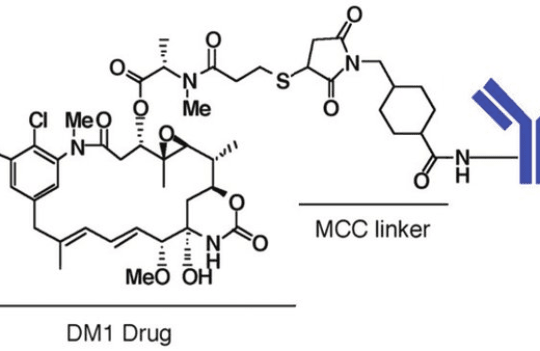
Early methods of antibody conjugation involved the reaction between an electrophilic group in the linker-payload complex with exposed amines (-NH2) in lysine residues of antibodies. This method of conjugation produced a heterogenous mixture of ADC species with variable drug loads.
Improvements in ADC homogeneity were subsequently achieved by partial or full reduction of functional groups available for reaction. This strategy limited the drug to antibody ratio (DAR) to 1-8. More recently, great progress has been made in site-specific conjugation methods. By engineering specific sites within the antibody, these conjugation methods have allowed the production of stable ADC with uniform species.
ADC conjugation methods can be classified as chemical or enzymatic.
Chemical conjugation relies on exposed and reactive functional groups of amino acid residues on the surface of the antibody fraction of an ADC. Both native or engineered (unnatural) amino acid residues can be used for conjugation including cysteine, lysine, and tyrosine, among others.
While lysine-based conjugation relies on reactive amine groups, cysteine conjugation methods require the reaction between these amino acid residues and a thiol functional group inserted on the payload. In human IgG1 antibodies, this is still the most commonly used method of conjugation. Exposed cysteine residues can be further blocked by cysteinylation or glutathionylation to limit the sites available for conjugation and thus achieve DAR with optimal therapeutic efficiency.
The use of unnatural amino acids is another strategy to allow better control over the site-directed conjugation process. This can be achieved by the introduction of unique codon-tRNA synthetases. These amino acids containing unusual functional groups such as carbonyl or azide provide more control over the conjugation between the antibody and its designed payload.
One of the most recent and exciting trends in antibody conjugation is the use of click chemistry. This approach represents a practical and highly efficient organic reaction that takes place under mild conditions and the presence of simple catalysts like copper. Click chemistry is quickly becoming the most popular and cost-efficient approach for antibody conjugation in ADC manufacturing due to tight control of conjugation sites and, subsequently, or DAR.
Some examples of ADCs in the clinical pipeline generated through click chemistry reactions include ARX788 (Ambrx), Trph-222 (Triphase), STRO-001 (Sutro Biopharma), and ADCT-601 (ADC Therapeutics), among others.
Many enzymes have been developed for the conjugation of natural and genetically engineered antibodies. Enzymes can modify the antibody in a site or sequence-specific manner. One approach to enzymatic antibody conjugation relies on the use of sortase A able to recognize a specific motif. This enzyme cleaves the threonine-glycine bond and attaches an oligoglycine molecule. Various cargos can be fused to this molecule including peptides, proteins, nucleic acids, and cytotoxic small drugs. For this reason, the use of sortase A remains a versatile and powerful approach to antibody conjugation.
Other enzyme-mediated strategies require the use of microbial transglutaminase or N-glycan engineering for site-specific integration. For instance, glycoengineering is particularly useful due to the presence of a conserved glycosylation site at position N297 in the CH2 domain. This site is distant from the antigen-binding sites ensuring that antibody affinity will not be altered by the process.
However, the efficiency of the process remains limited because glycosylation is a heterogeneous post-translational modification. Moreover, the modification of glycans may have the unintended effect of increasing antibody immunogenicity. Although studies have shown a better in vivo efficiency in comparison to lysine conjugates, this method requires more expensive reagents and enzymes for glycoengineering.
Antibody conjugation has a direct influence on the efficiency of ADCs. Controlling the number and site of conjugation is thus of vital importance. Modifications on specific amino acid residues or glycans are the two most popular approaches. Nevertheless, opting for amino acid-mediated conjugation is often the best approach considering the limited number of glycans on the antibody molecule and the time-consuming and labor-intensive process of glycoengineering.
After the selection of conjugation sites, it is necessary to determine if the number of sites available must be reduced to avoid overloading the antibody molecule. Moreover, it is essential to determine if the process of conjugation alters the binding affinity to the desired target.
Another aspect to control in this process is the stability of the conjugates. Stability is particularly important when developing ADCs with cytotoxic payloads for therapeutic applications. Stability requires the study of aggregation and degradation of ADCs after conjugation.
The coupling of large hydrophilic molecules (antibodies) with small hydrophobic molecules (linker-payload) remains challenging from a stability point of view. DMF (dimethylformamide) is often used to maintain the solubility of the hydrophobic counterpart of ADCs, but its presence is also known to promote excessive aggregation. One way to circumvent this issue is through the use of conjugation additives such as glycerol, propylene glycol, or octanoic acid.
Several methods can be used to measure stability and thus inform the process of conjugation for optimal results. These methods include Differential Scanning Calorimetry (DSC), Size-Exclusion Chromatography (SEC), Liquid Chromatography Mass Spectrometry (LC-MS), and electrophoresis. The results obtained from these measurements can be used to control the efficiency of the conjugation early on and allow the optimization of this process for optimal ADC efficiency.
The process of ADC conjugation is an active area of research. Several methodologies have been developed with varying degrees of success. The popular methods include amino acid-based conjugation and glycoengineering. In recent years, much progress has been made in the optimization of click chemistry reactions for optimal ADC conjugation.
Despite not being a new concept, click chemistry, fast organic reactions under mild conditions, has only been recently applied to ADC generation. This methodology ensures that the conjugation process remains simple and cost-effective while offering precise control over the number and position of conjugation sites.
The distribution of these sites influence the DAR and, subsequently, the stability and therapeutic efficacy of these conjugations. For this reason, controlling the efficiency of the process early on must go beyond the rational design of conjugation strategies. ADC stability is a good indicator of process efficiency, for this reason, methods for measuring this property should be used alongside ADC development.
You could also be interested in:
Join our email list to receive exclusive content featuring the most interesting industry and research news, biologics development tips pieced together by experts, res, company news, and exclusive limited-offers. Join a community of 80,000 subscribers and save up to 30% on your first order.