
Cart (0 Items)
Your cart is currently empty.
View ProductsIt looks like you are visiting from outside the EU. Switch to the US version to see local pricing in USD and local shipping.
Switch to US ($)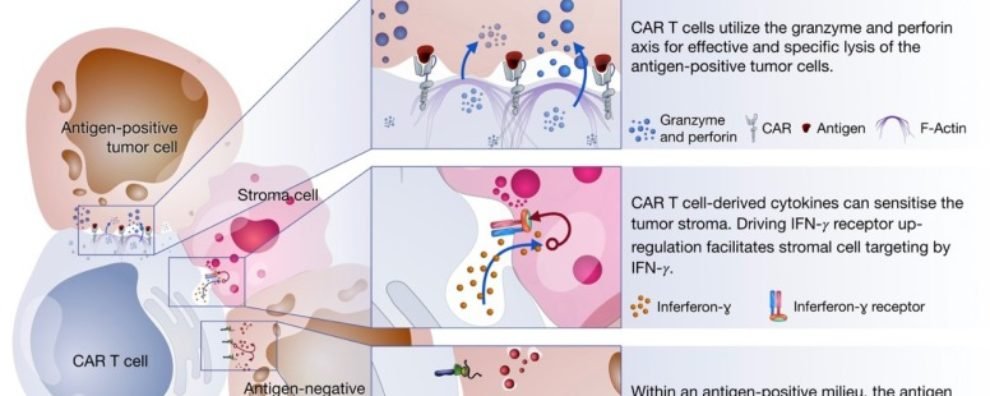 CAR-T cell therapy
CAR-T cell therapy
 Thomas Meyer
Thomas Meyer
Adoptive T cell therapy (ACT) is a type of immunotherapy that relies on the therapeutic use of T cells. ACT currently exists in three forms:
Of the three main forms of ACT, CAR-T cells remain the only form to have achieved significant clinical success to date. In contrast, TILs have found limited applicability due to the difficulties in harvesting lymphocytes from tumors and metastatic cancers. Moreover, since engineered TCRs are still dependant on MHC-mediated recognition and many tumors evade the immune response by losing or downregulating their MHCs, this approach remains very restricted.
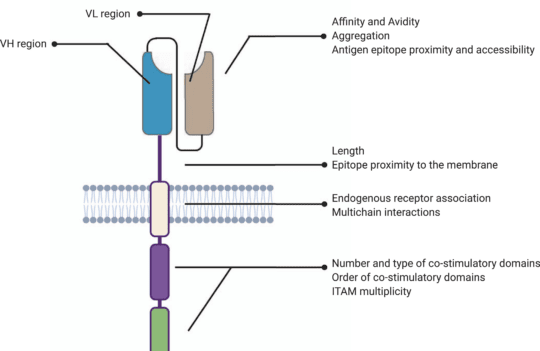
The hinge or spacer region is the extracellular structure of CARs extending from the antigen-binding domain to the transmembrane domain. This region lends flexibility to the scFv and can be used to enhance the recognition of target epitopes in otherwise sterically inaccessible regions.
Hinge design, namely length, and composition, also impacts CAR expression, signaling, and strength activation outputs. Optimal spacer lengths depend on the position of the target epitope and the level of steric hindrance on the targeted cells. For instance, numerous studies have shown that long spacers allow better access to membrane-proximal epitopes or glycosylated antigens. In contrast, short hinges are more useful when targeting membrane-distal epitopes. But, in practice, spacer length needs to be tailored for each specific ligand-scFv pair. Similar to scFv affinity towards specific antigens, long spacers can produce particularly strong in vivo responses but also lead to activation-induced cell death.
Most commonly used hinge regions are derived from membrane-bound receptors (CD8 or CD28) or IgG (IgG1 or IgG4). However, IgG-derived spacers are known to interact with Fcγ receptors (FcγR), leading to the activation of the innate immune response and CAR-T cell depletion. For this reason, most IgG-derived spacers are mutated to minimize interactions with FcγR.
Besides their role in CAR signaling, hinge regions are commonly used to quantify and purify CAR-positive subsets of T cells after engineering. For this purpose, anti-Fc antibodies can be used when working with IgG-derived spacers. Moreover, Strep-tag sequences have been successfully introduced in the CAR spacer region to ensure a more efficient quantification and purification of CAR-positive cells.
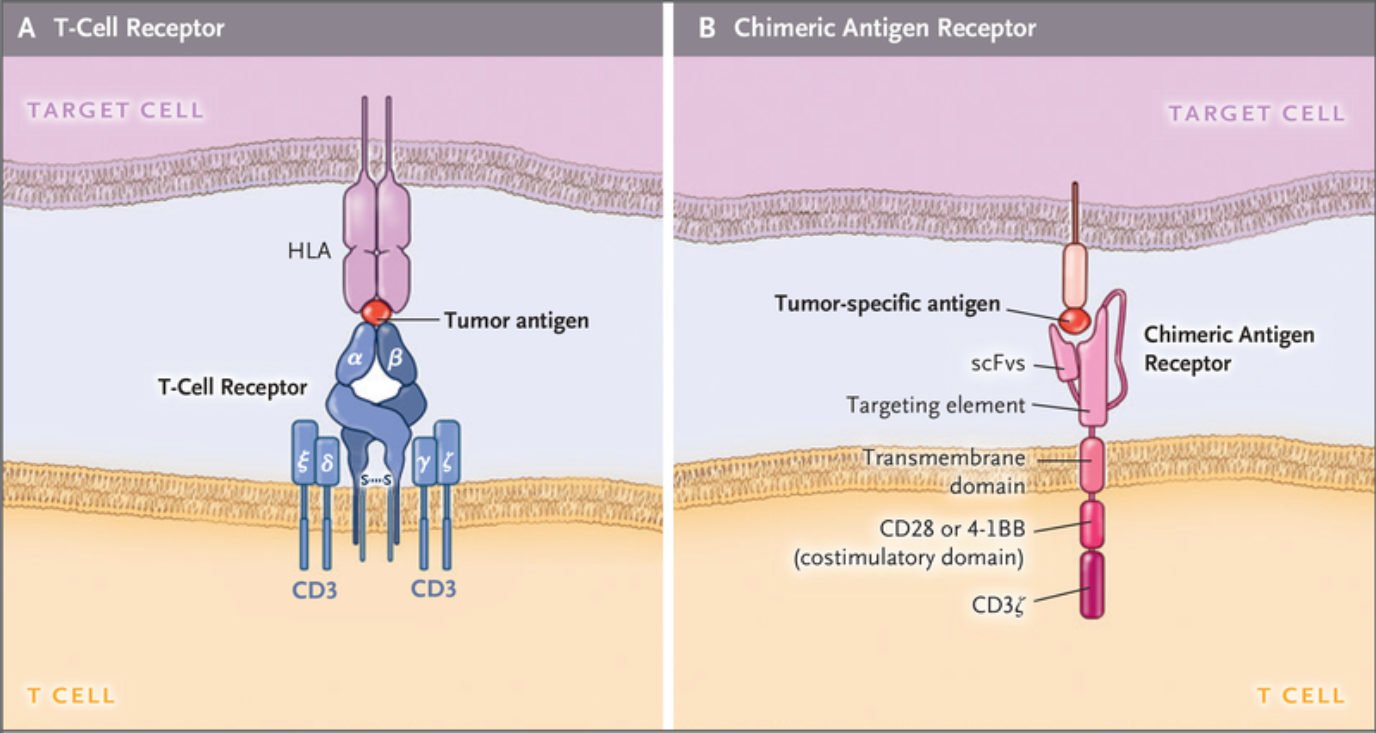
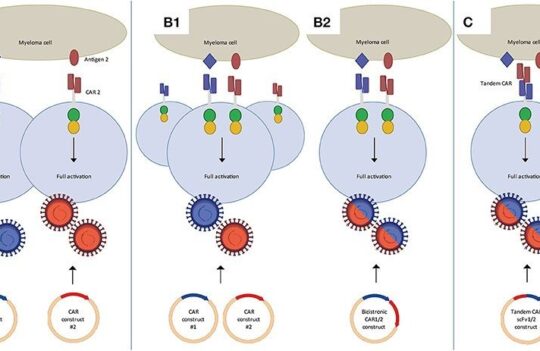
CARs represent the most complex form of ACT. They are synthetic structures designed to mimic TCRs. They share similarities in terms of signaling pathways, but the fundamental difference between the two lies in the fact that CARs possess an engineered scFv domain able to recognize tumor antigens not bound by MHC molecules and, instead, expressed directly on the membrane. In this way, unlike engineered TCRs, CARs can target tumor cells that have lost or downregulated their MHCs (immune escapes).
However, the ability to target antigens in an MHC-independent way creates another challenge because these modified cells lose the ability to target intracellular disease markers. This poses an important limitation as much of the work done on cancer biomarkers has focused on intracellular targets. The limited knowledge regarding tumor-associated membrane-bound antigens explains why all currently approved cell therapies target CD19. Going beyond this conventional target is thus of vital importance but it may require looking past protein receptors and into glycolipids or other surface-bound components of the membrane.
Despite their structural differences, CARs and TCRs share an essential signaling component – a CD3ζ chain. This chain contains 3 immuno-tyrosine activation motifs (ITAMs) that participate in the cell signaling process. When CARs or TCRs encounter an antigen, the event triggers the phosphorylation of ITAMs via the lymphocyte-specific protein tyrosine kinase (Lck). In first-generation CARs, a single CD3ζ chain was the only intracellular domain.
However, these simple synthetic receptors showed low cytotoxicity and proliferation. Their limited response was attributed to the lack of costimulatory domains (CD28 or 4-1BB), found to be instrumental for sustaining the signaling cascade and triggering the production of cytokines. For this reason, newer generation CAR-T cells contain costimulatory domains adjacent to the CD3ζ chain. Moreover, latest generation CAR-T cells may contain additional inducible transgenes encoding for secreted molecules (e.g., IL-12) or membrane receptors, designed to enhance the proliferation of these “living drugs.”
Natural cytotoxic T cells (CTLs comprise CD4+ and CD8+ T cells) eliminate cancerous or infected cells with remarkable specificity. The formation of a stable immune synapse (interface between receptor and ligand) is the main triggering factor of this response. For a long time, researchers believed CAR-T cells formed a comparable immune synapse because they shared part of their signaling domains with endogenous TCRs. However, recent studies showed that a CAR-mediated immune synapse is significantly different from TCR-mediated responses. In comparison, CARs generate stronger signals quicker than TCRs, but their cytotoxic response appears to be comparable.
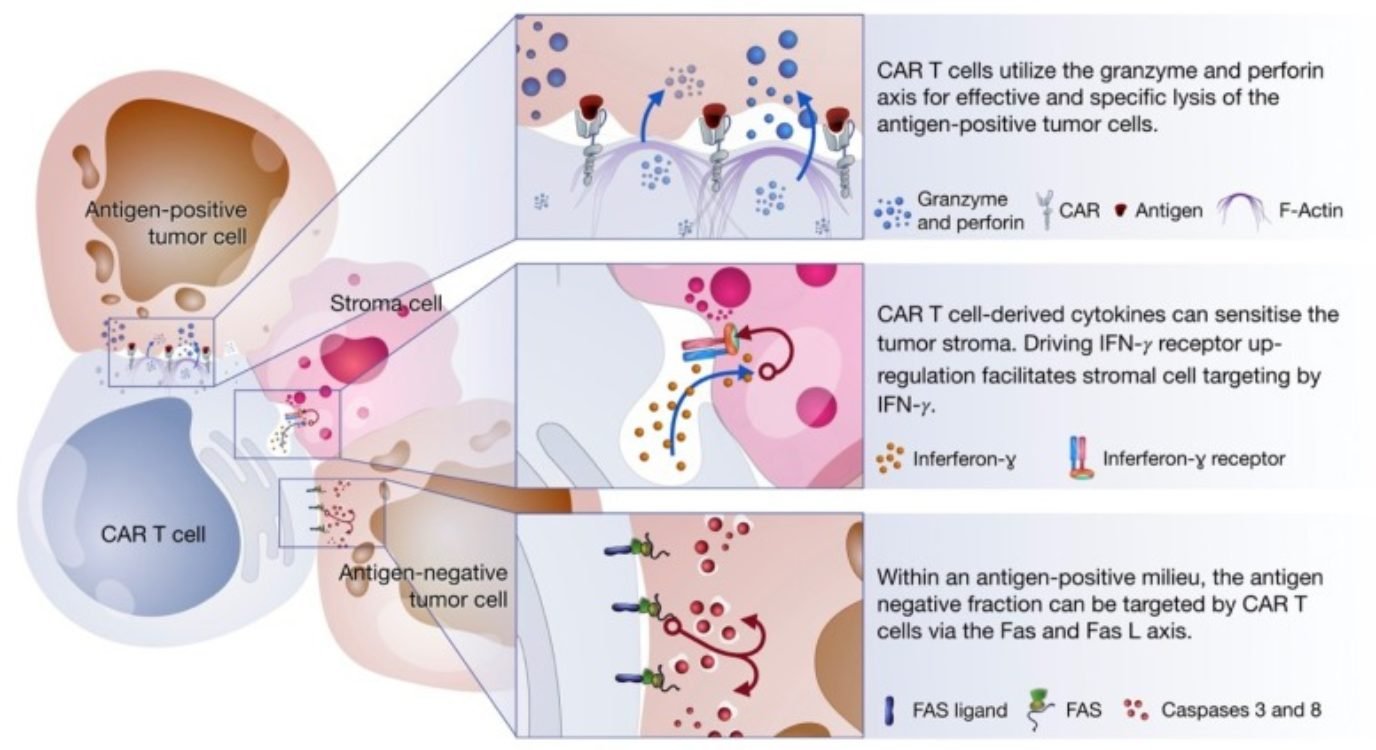
The cytotoxic response of CAR-T cells occurs via three axes:
All these mechanisms are triggered upon encounter with the tumor-associated antigen. Interestingly, they target different cell types within the tumor milieu.
Degranulation is thought to be the main cell killing mechanism of CAR-T cells. It takes place after the establishment of the immune synapse with the tumor cells. This response mostly occurs via the delivery of cytotoxic granules containing perforin and granzymes. The granules migrate towards the interface between T cell and cancer cell where perforins induce pore formation facilitating the access of granzymes to cancer cells. Once granzymes reach the cytoplasm, they are free to induce apoptotic cell death in a caspase-dependent or independent way.
The Fas-FasL is a major apoptosis pathway via caspase-dependent activation. Interestingly, this mechanism is only activated once CARs bind their specific antigen, but it was shown to mediate the lysis of antigen-negative cells in the tumor milieu. Since cancer cells are prone to antigen loss and tumors are heterogeneous by nature, this alternative mechanism remains extremely important to increase efficacy and avoid disease relapse due to antigen escape.
In comparison to the two previous mechanisms, cytokine production by activated CAR-T cells induces cell death via secondary mechanisms of action. Cytokines trigger several anti-tumor immune responses including the enhancement of the cytotoxic response, recruitment and activation of innate immune cells (i.e., macrophages, NK cells, etc.), and reprogramming of stroma-associated immune suppressor cells by upregulating IFNɣ receptor expression. Stroma is present on solid tumors and mostly consists of connective tissue and blood vessels acting as a physical barrier that blocks access to tumor cells. Studies show that stroma targeting is crucial to prevent relapse and allow the complete remission of cancer. For this reason, the sensitization of stroma cells is thought to be an important mechanism of elimination employed by CAR-T cell therapies.
The first time a CAR-T cell therapy was successfully used to treat a form of cancer was in 2010. Within 7 years, the Food and Drug Administration (FDA) approved the first two CAR-T cell therapies – KymriahTM and YescartaTM. Since then, a total of five of these therapeutics have reached the market. Three of these have also received marketing authorization for the European Medicines Agency (EMA).
| Name | Target | Clinical indication | Date of approval (FDA) |
|---|---|---|---|
| Abecma® (idecabtagene vicleucel) | TNFRSF17 (TNF receptor superfamily member 17, BCMA, tumor necrosis factor receptor superfamily, member 17, BCM, CD269, TNFRSF13A) | Multiple myeloma (MM) | March 2021 |
| Breyanzi® (lisocabtagene maraleucel) | CD19 (B lymphocyte surface antigen B4, Leu-12) | Lymphoma, diffuse large B cell (DLBCL) | February 2021 |
| KymriahTM (tisagenlecleucel) | CD19 (B lymphocyte surface antigen B4, Leu-12) | Acute lymphocytic leukemia (ALL) | August 2017 |
| TecartusTM (brexucabtagene autoleucel) | CD19 (B lymphocyte surface antigen B4, Leu-12) | Mantle cell lymphoma | July 2020 |
| YescartaTM (axicabtagene ciloleucel) | CD19 (B lymphocyte surface antigen B4, Leu-12) | Lymphoma, diffuse large B cell (DLBCL) | October 2017 |

New perspectives for Car-T therapy: Explore how we designed antibodies against a target barely visible to the immune system
Read the full Case ReportCAR-T cell therapies combine the cytotoxic potential of T cells with the specificity of antibodies in the form scFv domains. Currently, five of these emerging therapies have received marketing approval and most have been designed to target B cell malignancies via CD19. Interestingly, after less than five years since the approval of the first CAR-T cell therapy, dozens of others are already undergoing phase 3 or 4 clinical trials.
Despite the challenges, CAR-T cells have found success in the treatment of refractory tumors, particularly those that remain unresponsive to conventional therapies. These cells were shown to cause the lysis of tumors in an efficient and specific way making use of several mechanisms such as degranulation, Fas-FasL pathway, and cytokine production.
In the next decades, CAR-T cells are expected to become safer, more effective, and applicable to a vaster range of oncological and non-oncological diseases.
Join our email list to receive exclusive content featuring the most interesting industry and research news, biologics development tips pieced together by experts, res, company news, and exclusive limited-offers. Join a community of 80,000 subscribers and save up to 30% on your first order.