
Cart (0 Items)
Your cart is currently empty.
View ProductsIt looks like you are visiting from outside the EU. Switch to the US version to see local pricing in USD and local shipping.
Switch to US ($)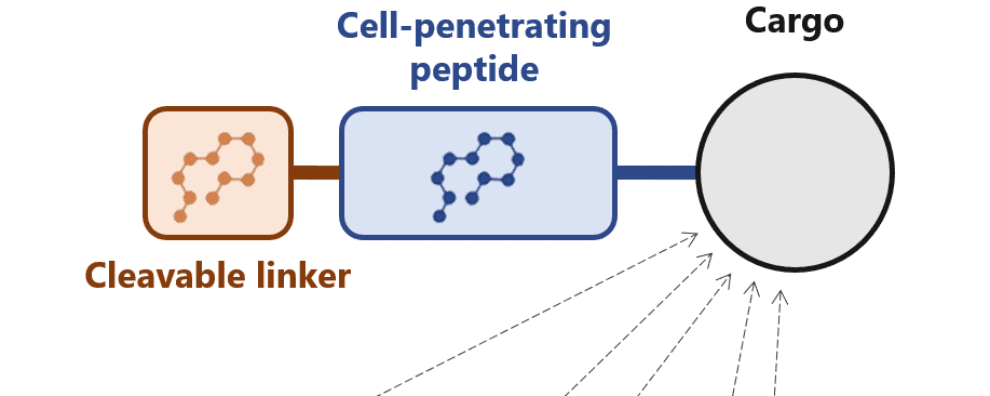 Peptide synthesis
Peptide synthesis
 Thomas Meyer
Thomas Meyer
On a molecular level, peptides are poised between small and large molecules, consisting of short chains of 2-150 amino acid residues linked by peptide (amide) bonds.
They are naturally widespread and ubiquitous molecules, playing essential roles in the regulation of the human metabolism and as signaling molecules across all domains of life. Due to their widespread nature, there are obvious advantages in harnessing the potential of these molecules for therapy. Contrasting with small chemical molecules, peptides can mimic natural ligands and interact with natural enzymatic or cell receptors. They are also highly specific and easy to produce, which makes them ideal candidates for a smooth transition from research to the clinic.
Moreover, peptides can be easily cleaved by naturally occurring proteases, releasing amino acids that can be recycled by our organism through multiple metabolic pathways. Therefore, reducing the risks of long-term side effects of peptide-based therapies.
But the history of peptides and their subsequent development for therapeutic applications is not without obstacles and short-comings.
The central obstacle for the application of peptides in therapy stems from their very nature. Native peptides are naturally hydrophilic and easily degraded. And although this is often an advantage for multiple applications, it limits the use of these biomolecules when it comes to therapy.
This happens because the usefulness of a therapeutic agent is often defined by their inherent stability and ability to reach a specific target. For the treatment of many human pathologies, including cancer and central nervous system (CNS) diseases, drugs need to cross highly hydrophobic lipid-rich membranes, impermeable to native peptides.
Peptides are also natural nutrients and thus are promptly digested by proteases upon oral administration. Moreover, these molecules lack tissue specificity, which further limits their application for the treatment of many tissue-specific pathologies.
Opportunities to bypass the limited diffusion of peptides have started to gain shape after the discovery of translocations capacities in native proteins. The first two proteins reported to have this ability were described in the late 1980s and early 1990s by three independent research groups. The first protein to be described was the trans-activator of transcription (TAT) protein of HIV-1. This protein and its properties were first described by researchers from John Hopkins University and Saint Louis University in the US who found that TAT could quickly be internalized by cells in vitro. The second protein was the homeotic protein antennapedia from Drosophila melanogaster described by researchers from the Ecole Normale Supérieure in Paris, France.
The discovery of these proteins was quickly followed by further studies that led to the identification of the specific peptide domains responsible for the cellular uptake. The characterization and isolation of these domains further led to the identification of many other peptides with translocation abilities.
Collectively these molecules were defined as cell-penetrating peptides.
They usually consist of 5 to 30 amino acid residues that can translocate through tissues and membranes. The diffusion of these peptides across membranes, also known as membrane transduction, is still not fully understood. But it is known that it occurs through energy-independent mechanisms either by direct penetration or by the endocytosis pathway.
Both pathways involve the initial interaction of the positively charged peptides with the negatively charged membrane components. This initial contact is followed by a complex process causing transient pore formation or endocytic vesicles, membrane destabilization and subsequent internalization of these molecules.
At this point, another limitation of using peptides for therapy persisted: its lack of tissue specificity.
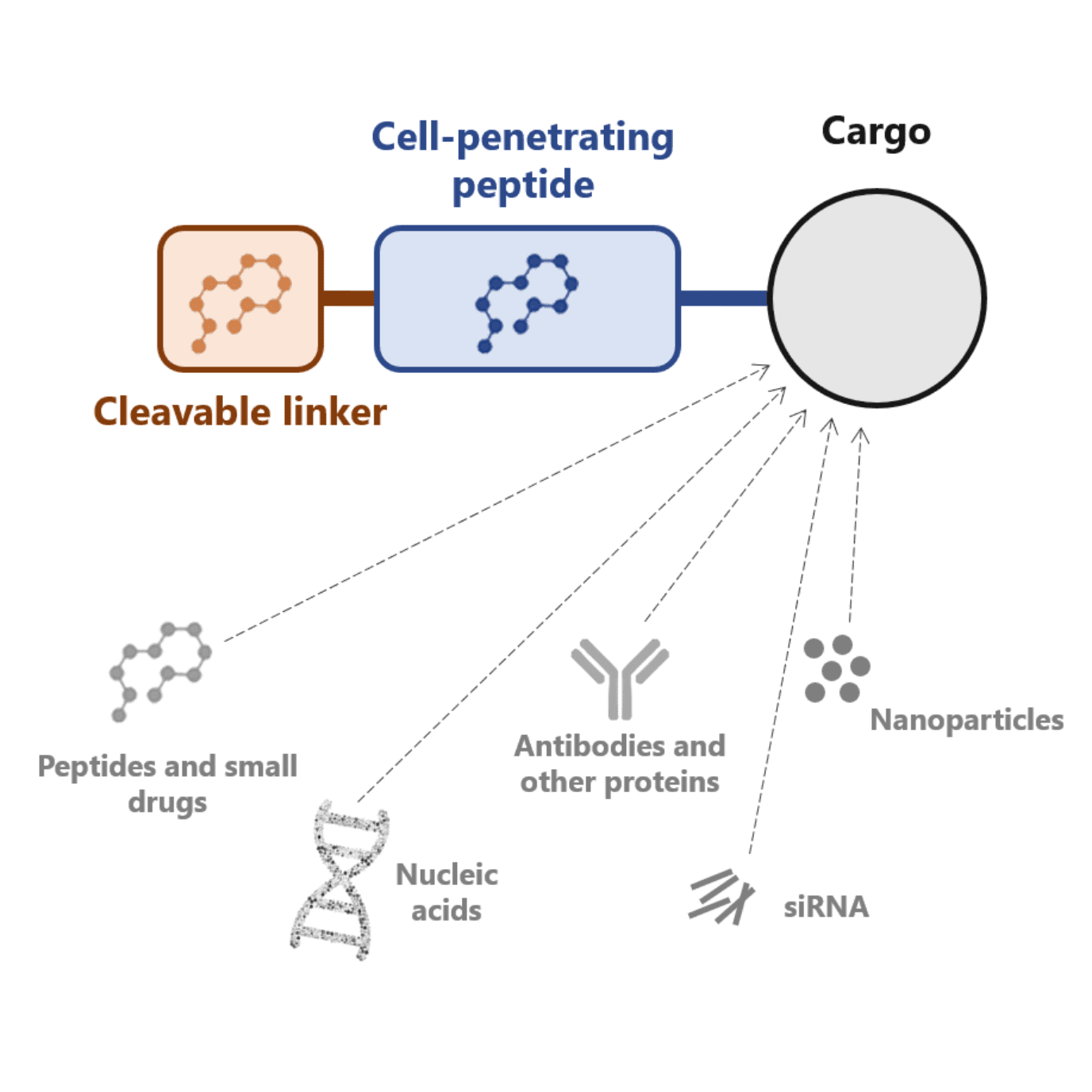
Several approaches have been developed to overcome this limitation. One of these approaches consists of conjugating cell-penetrating peptides with antibodies. Another potential solution consists of masking these peptides with a cleavable linker.
The latter was first proposed by researchers from the University of California, who named these peptides activatable cell-penetrating peptides. They fused a commonly used peptide with a negatively charged amino acid chain that blocked the uptake of the molecule. Interestingly, this linker had been designed to be cleaved by tissue-specific proteases, resulting in an elegant solution for the delivery of biological cargoes to specific tissues.
These translocating peptides can be classified and grouped according to their physicochemical properties and defined as cationic, amphipathic or hydrophobic. A few examples of peptides belonging to each specific class can be found in the table below:
| Name | Amino acid sequence (residues) | Class | Description |
|---|---|---|---|
| HIV-1 TAT protein (TAT49-57) | RKKRRQRRR (9) | Cationic | Possessing highly positive net charges at physiological pH. It is estimated that arginine residues (R) are of extreme importance for their internalization. |
| Penetratin (pAntp43-58) | RQIKIWFQNRRMKWKK (16) | ||
| DPV1047 | VKRGLKLRHVRPRVTRMDV (19) | ||
| Pep-1 | KETWWETWWTEWSQPKKKRKV (21) | Amphipathic | Containing both polar (hydrophilic) and nonpolar (hydrophobic) regions. |
| pVEC | LLIILRRRIRKQAHAHSK (18) | ||
| MAP | KLALKLALKALKAALKLA (18) | ||
| C105Y | CSIPPEVKFNKPFVYLI (17) | Hydrophobic | Consisting mainly of nonpolar residues, which results in a low net charge. |
| PFVYLI | PFVYLI (6) | ||
| Pep-7 | SDLWEMMMVSLACQY (15) |
Independently of their physiochemical properties, cell-penetrating peptides can be conjugated with different cargoes (e.g. peptides, proteins and small molecules). A conjugation that can be made via covalent and non-covalent binding.
Covalent binding can be achieved by chemical conjugation or cloning and subsequent expression of the fused peptide-cargo conjugate. However, this linkage often takes the form of disulfide or thioester bonds which can significantly alter the biological activity of the conjugates.
Hence, non-covalent binding is often considered a better alternative for the production of these conjugates. This type of binding relies on electrostatic, hydrophobic interactions or a combination of the two between negatively charged cargoes and positively charged cell-penetrating peptides. Besides preserving the activity of the conjugates, this type of interaction also protects the conjugates against protease and nuclease degradation, further increasing their half-life.
Cell-penetrating peptides can be conjugated with a plethora of therapeutically valuable molecules such as:
Multiple cell-penetrating peptides and conjugates thereof are currently under preclinical and clinical trials. Their ability to transport biologically active cargoes across membranes has revived the interest of industries and researchers in other therapeutic molecules that have previously faced enormous challenges in the transition to the clinic, such as gene and siRNA therapies.
These molecules, although presenting good activity in vitro, were found to suffer from one important drawback: their inability to cross membranes and reach the desired target.
The discovery of versatile cell-penetrating peptide continues to prompt and enhance the research on these conjugates as an elegant way to overcome this central problem of modern medicine.
Join our email list to receive exclusive content featuring the most interesting industry and research news, biologics development tips pieced together by experts, res, company news, and exclusive limited-offers. Join a community of 80,000 subscribers and save up to 30% on your first order.