


Cart (0 Items)
Your cart is currently empty.
View ProductsIt looks like you are visiting from outside the EU. Switch to the US version to see local pricing in USD and local shipping.
Switch to US ($) Antibody production
Antibody production
 Thomas Meyer
Thomas Meyer

Dr. Mohamed Alfaleh: I started my journey by enrolling and graduating from a bachelor’s in pharmacy at the top of my class. This allowed me to obtain a scholarship that I used to pursue my master’s and PhD studies in biotechnology at the University of Queensland (Brisbane, Australia). I later returned to King Abdulaziz University (KAU) where I became an assistant professor. I wanted to be a scientist for as long as I can remember, but I landed on this field of antibody development by phage display mostly due to an opportune coincidence.
When I was looking for a PhD position, I had the opportunity to attend a lecture about antibody technologies. This lecture was impacted by Professor Stephen M. Mahler who was part of the original team leading the efforts that culminated in the discovery of Humira® (Adalimumab, the first phage display-derived antibody granted marketing approval) (doi: 10.1038/nbt0994-899). Learning from the experience of earlier generations inspired me to pursue my PhD in this field and led me to join Prof. Mahler’s lab, where my journey in antibody phage display truly begun.
Hybridoma technologies were ground-breaking at the time and they are still commonly used to produce antibodies for research. But mice-derived murine antibodies have severe side effects. These effects are caused by human anti-mouse antibody (HAMA) responses, which accelerate antibody clearance and can result in serious immunogenic reactions.
Antibody engineering techniques were initially devised to minimize these adverse reactions. Soon after the discovery of murine antibodies and the discovery of the HAMA response, antibody engineering soon allowed us to modify the original murine molecule into chimeric and humanized antibody variants, but these solutions are still far from perfect.
In contrast, transgenic mice models can be coupled with hybridoma technologies to generate fully human antibodies. But phage display still has an impressive advantage over these technologies because it allows us to bypass animal immunization and still produce fully human antibodies in different formats (mAb or single-chain formats). But the most important advantage of phage display over hybridoma generation in transgenic mice is the compatibility of this technique with the use of toxic or non-immunogenic antigens, which cannot be generated in animal models. Additionally, phage display allows us to isolate conformation-specific antibodies because you have the luxury of manipulating the antigen at specific conformations.
This preference for hybridoma-derived antibodies happens because mammalian cells are more efficient in performing affinity maturation. This process then results in antibodies with higher affinities than the ones currently obtained by phage display technologies.
Plus, these mice-derived antibodies generally have better biophysical attributes, which are vital determinants of the antibody’s developability for biomanufacturing. It has been found that antibodies derived from in vitro selection tend to have high aggregation rates and polyspecificity. This undesirable behavior results from the favored selection of hydrophobic residues in the CDRs (complementary determining regions) in conventional phage display technologies.
This can be solved by developing higher quality libraries with greater diversity and coupling them with improved panning procedures. Others have also reported an increase in specific libraries’ developability by testing antibodies at high temperatures because high thermal stability is crucial to maintain antibodies’ structural and functional integrity and avoid long-term aggregation. With the booming of bioinformatics tools for antibody assessment, it is now also possible to determine an antibody potential developability using only its sequence (e.g. Therapeutic Antibody Profiler, TAP).
Membrane proteins are desirable targets for many applications. They account for 20 to 30% of all biologically relevant proteins and represent about 44% of all commonly used targets for drug discovery. But despite the huge progress made in protein engineering and manufacturing, membrane protein expression and purification can still be quite challenging and time-consuming.
For some membrane-bound proteins, it is simple to just purify the extracellular domain and use it to obtain high-affinity antibodies that are also active against native conformations. But for many other proteins, such as ion-channels or G protein-coupled receptors, the large-scale expression is quite tricky, and native conformations are easily lost outside the original hydrophobic environment.
If we perform panning against these unusual conformations, we risk isolating antibodies against epitopes that are either denatured or not naturally exposed in the native protein, and thus biologically irrelevant. Cell-based phage display can overcome these problems because the antigens are presented in their native or near-native conformations with appropriate post-translational modifications, which heavily influence antigen-antibody interactions.
Another lesser-known advantage of cell-based over purified protein-based biopanning is the possibility to isolate antibodies against unknown or uncharacterized epitopes present on the surface of specific cell populations. This gives us the unique possibility to further characterize these populations and uncover new biologically relevant targets for antibody development.
This is the first and most critical step in any cell-based phage display project. Some researchers use native cell lines, which present numerous advantages in terms of structure and post-translational modifications, but are still extremely challenging to work with. In these cases, low receptor density, high background noise (due to the presence of carbohydrates, lipids, unspecific proteins), and unspecific adsorption remain the most important technical constraints.
On the contrary, the use of engineered cell lines gives us a greater degree of control. Starting from CHO (Chinese hamster ovary) or HEK (Human embryonic kidney) cell lines allow us to exert more precise control over antigen density. This can be done by fusing the antigen of interest to GFP (green fluorescent protein) and then analyzing and separating the high-intensity cells with FACS (fluorescence-activated cell sorting). The use of engineered cell lines enables us to reduce unspecific binding by performing strategic subtractions with alternative rounds of panning with transfected or untransfected CHO and HEK cell lines.
The use of CHO or HEK modified cell lines also gives us an important advantage. Unlike we saw with some native cells, the use of CHO and HEK allows us to perform suspension-based cell panning. Because interestingly, performing panning on confluent monolayer cells (e.g. epithelial cells) was found to increase the risk of isolating antibodies against the plastic surface containing the cells or the serum proteins found in the culture medium. We can easily avoid these issues by doing suspension-based cell panning. Additionally, by performing panning in suspension we can increase the surface area with our target antigen.
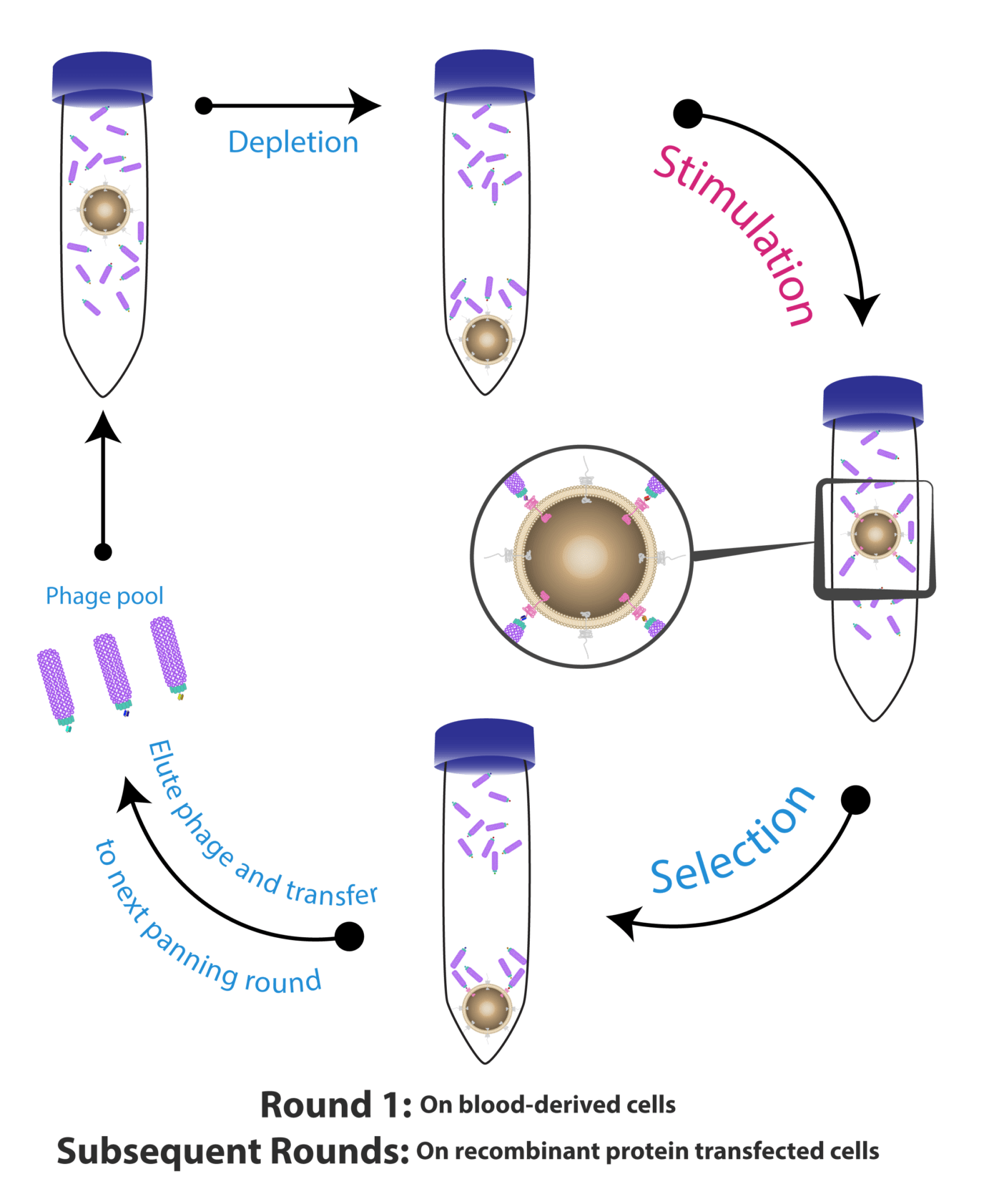
On a more technical aspect, we found that fine-tuning some conditions allow us to recurrently obtain better performances. For instance, performing washes at low pH (5) allows us to significantly reduce the abundance of phages carrying no binders or unspecific binders. The concentration of Tween (essential detergents to avoid cell aggregation) also heavily influences antibody specificity and should be carefully optimized for each project.
At the same time, it is also important to make this harsh wash protocol as elegant as possible to avoid destroying the cells and thus leak irrelevant intracellular antigens into the medium. Moreover, incubating the cell and phage mixture at 37°C was shown to cause phage internalization which is highly undesirable. This can be avoided by incubating the mixtures at low temperatures for short periods (4°C for 2h) to avoid binders’ internalization and desensitize cells from environmental stimuli.
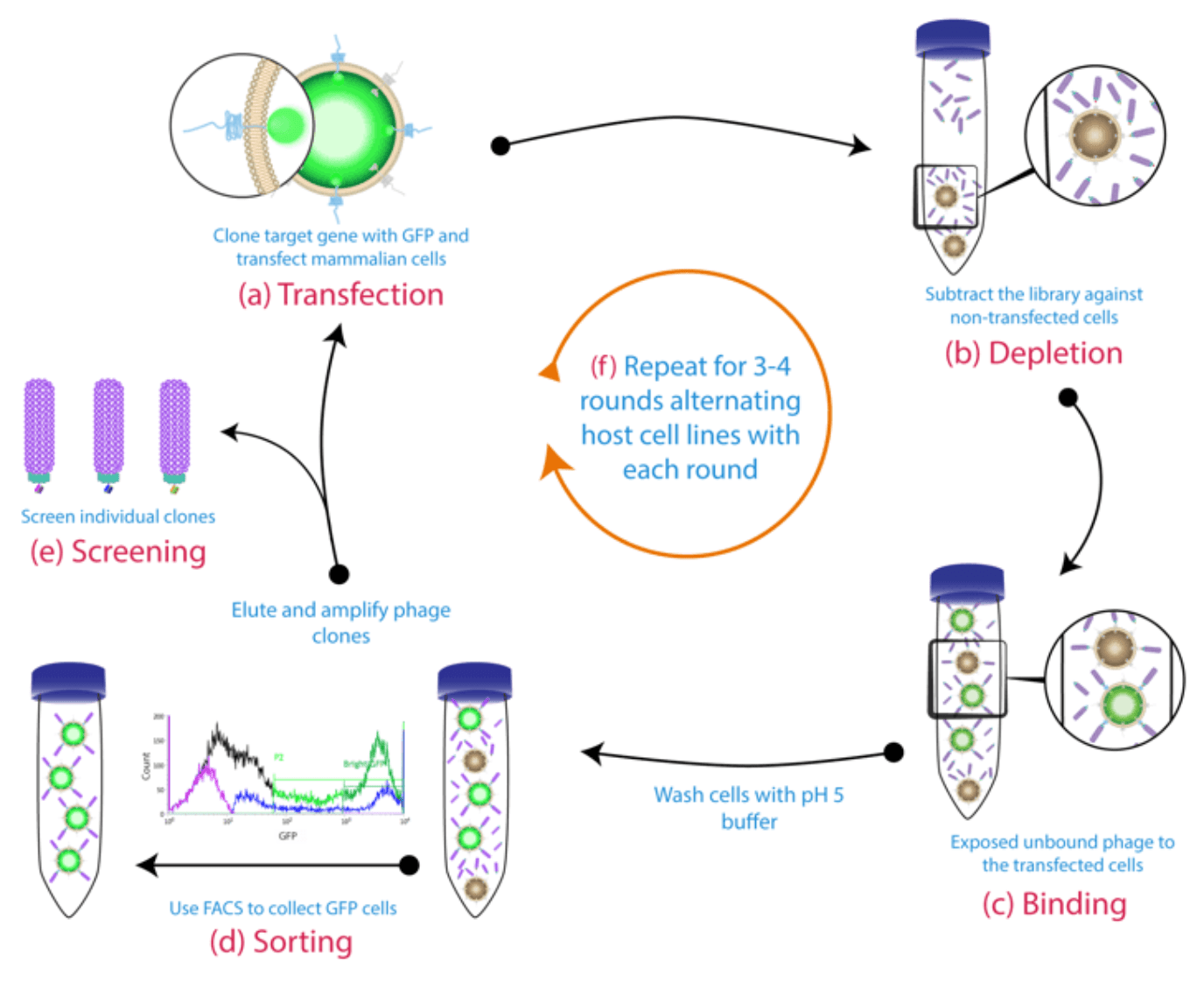
Cell-based phage display is not yet a plug and play type of technology; it must be optimized in a case-by-case approach which makes this approach incompatible with projects evaluating multiple antigens or projects requiring fast turnovers. But despite these limitations, cell-based panning is still an invaluable methodology to generate antibodies against complex native conformations, such as antibody drugs or antibody markers used in clinical diagnostics.
Surprisingly, one of our most important discoveries was characterizing these mAbs and showing their binding properties were quite diverse, despite being isolated during the same cell-based biopanning campaign. The reactivity of the isolated antibodies varied between the assays and the antigen source.
Although it was useful to validate the isolated antibodies against soluble recombinant forms of target antigens for specificity and affinity, this did not necessarily represent antibody-antigen interactions in their natural context. These outcomes demonstrated the power of the cell-based biopanning method, which allowed us to isolate a panel of mAbs that could access the native antigen and bind to different epitopes.
For instance, biopanning against whole cells may lead to binders that bind weakly in ELISA and SPR (surface plasmon resonance) but bind well to the native antigen with flow cytometry. Therefore, we recommend that during whole cell-based biopanning campaigns, all isolated antibodies should be included in all assessments without exclusion, even those antibodies that seem unpromising during the initial analysis. It is essential to validate the isolated mAbs on a soluble recombinant form of the targeted antigen to confirm the specificity and assess the binding affinity and avidity, yet this does not necessarily represent what is naturally occurring between the antibody paratopes and the antigen’s epitopes.
In Australia, malignant mesothelioma is very prevalent due to the presence of asbestos which were immensely popular during World War I and still predominant in many old and heritage buildings. Prolonged exposure to asbestos is known to increase the risk of developing mesothelioma, for which the symptoms may only manifest 20-50 years after exposure. At this point, tumors have already grown and spread, and, with current treatments, patients are only expected to live 12 to 22 months after the initial diagnosis. This work represents a steppingstone in the development of better treatments for this prevalent disease. And it resulted from a long-term collaboration between the University of Queensland’s Australian Institute for Bioengineering and Nanotechnology (AIBN) and EnGeneIC Ltd.
The nanocells used in this study were originally obtained from Salmonella typhimurium and they carry a surprisingly high concentration of drugs (up to 10 million molecules per nanocell). Prior studies published by our group revealed that the best bispecific format for delivering these drug-charged nanocells was the tandem single-chain bispecific antibody format (scFv-BsAb). Due to the bivalent nature of these antibodies, we can design one arm to target the cancer antigen (mesothelin) and the other against the bacterial LPS (lipopolysaccharides) present in the nanocells.
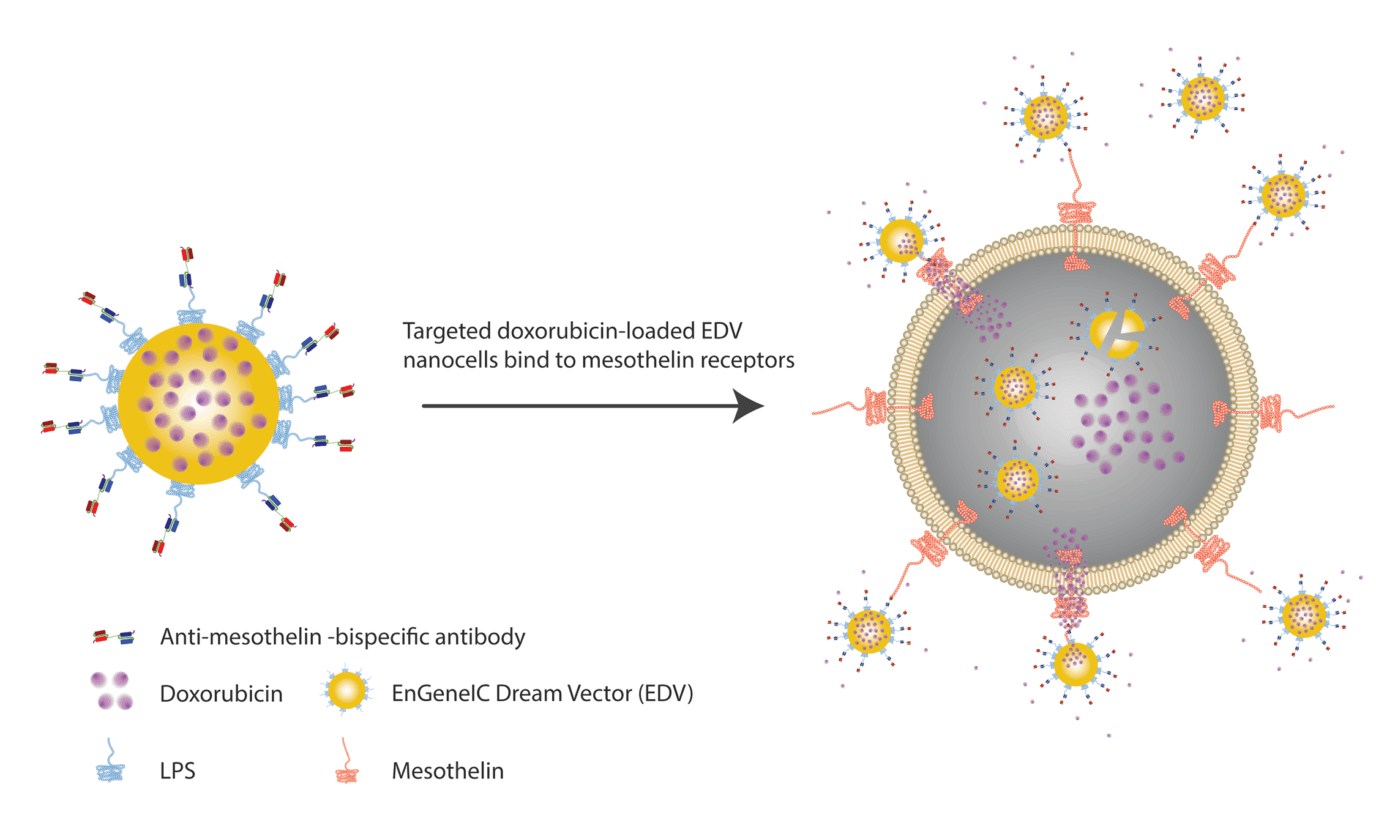
In our study, we used Amatuximab (a phage display-derived monoclonal antibody) and reformatted it to a single-chain bispecific format. Our in vitro, in vivo, and ex vivo studies with mouse xenografts all revealed that the targeted delivery of these nanocells reduced tumor proliferation and enhanced apoptosis in animal models.
Like most biopharmaceuticals, large-scale production might be challenging. On the one hand, it is necessary to deal with the typical challenges imposed by bispecific antibody production and, on the other hand, it is necessary to optimize the production and purification of bacterially derived nanocells. And this process must be done in tandem before the final assessment of the construct’s stability.
We found significant variations between different commercially available serological tests. We believe these variations can be explained by insufficient validation and optimization, which are crucial to improve the usefulness of these tests for large-scale assessments.
We developed an in-house ELISA test to detect the levels of antibodies against SARS-CoV-2 using both the S1 (subunit 1 of the spike protein) and the N (nucleoprotein) as markers. The most challenging part of the study was finding a suitable cutoff to differentiate between positive and false-positive signals. But once we were able to identify this cutoff it was easy to validate the test. From there on, these tests can be used to assess the prevalence of infection among the population.
Unlike RT-PCR, which measures the presence of the virus, ELISA evaluates immunity. For this reason, ELISA is more useful to perform an early assessment of the efficiency of different vaccination strategies and, in general, to provide a broader picture of how immunity to SARS-CoV-2 evolves within a specific population.
Our group is primarily focused on developing new vaccines, and this is currently being pursued using machine learning algorithms and artificial intelligence. In our region, the field of antibody development is still pristine. Thus, our primary and initial focus is to build capacity. At the same time, we are planning on pursuing challenging projects in antibody library development and improve current discovery, engineering, and production methods for bispecific antibodies.
The production of bispecific antibodies remains quite challenging because, unlike monoclonal antibodies, bispecific production typically yields a heterogeneous mixture of different combinations of light and heavy chains. Presently, a typical purity ratio for bispecific antibodies is somewhere between 50 and 25%. But this may soon change thanks to the use of phage display for bispecific antibody engineering.
The proof of concept was recently published by Prof. Daniel Christ’s group at the Garvan Institute of Medical Research (Sydney, Australia) (doi: 10.1021/acs.biochem.9b00037). They developed a phage display-based methodology to improve the affinity between the heavy and light chains. In this way, instead of the typical yields, this new methodology allowed them to obtain purity ratios above 80%. And this is something that I would also like to pursue further in the future.
As Prof. Christ recently showed, phage display will likely be increasingly used for antibody engineering. Also, we will likely soon witness the development of more efficient high throughput systems for phage display-guided antibody discovery, such as the use of nanotechnology and next-generation long-read sequencing to accelerate current panning protocols.
In the coming decades, it is also probably we will see an increase in the number of studies dealing with alternative display methods such as yeast and mammalian cell display. I believe we will see the greatest breakthroughs in mammalian cell display, because these systems are uniquely fitted to produce antibodies with proper glycosylation profiles, and they can display them in full monoclonal conformations. This would give us unique and early insights into antibody developability and binding affinity.
Join our email list to receive exclusive content featuring the most interesting industry and research news, biologics development tips pieced together by experts, res, company news, and exclusive limited-offers. Join a community of 80,000 subscribers and save up to 30% on your first order.